Introduction
Vision is one of the five senses the body uses to interpret its surroundings. In the past, our primitive ancestors had what is called "dichromatic vision,” allowing for interpretation of only UV light and red light. About 30 million years ago, the trichromatic part of vision came to existence due to the evolution of opsin genes.[1] Humans can now see black, white, red, green, and blue, as well as the colors in between this spectrum since the retina and the brain are equipped to differentiate them. What happens between an object and a synapse in the most posterior part of the brain is the fascinating journey that we will cover.
Issues of Concern
Physical Properties of Vision
Vision cannot be discussed without knowing the physical properties of optics. The eye receives light that then is traduced into energy. That energy goes into the optic nerve as an action potential and travels to specific nuclei in the brain, where it is processed. But, how does that light get into the eye to be processed into an action potential to be sent to the brain?
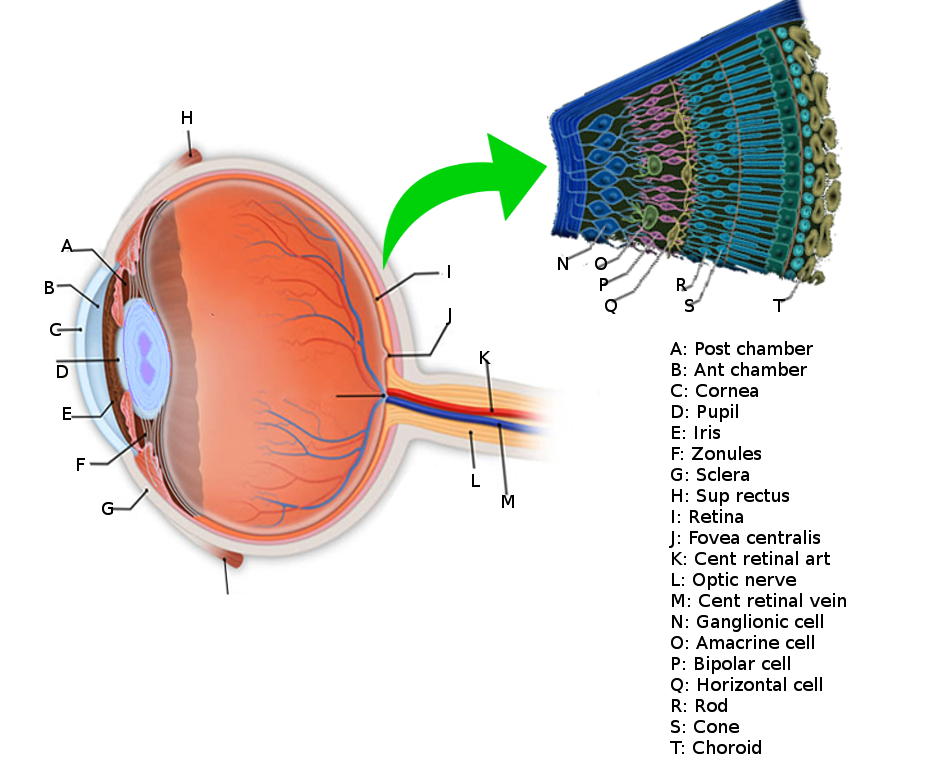
The eye is composed of a series of lenses and spaces that give focus to images, just as a camera does. It is composed of the vitreous humor, aqueous humor, the crystalline lens, and the cornea, and each of these has its own refraction index (the average being 1.34, because of the content of these tissues). Light travels in the air in the form of waves. The term “refraction index” refers to the relation between the velocity of light in the air compared to its velocity when it travels across an object. Light travels at a velocity of 300,000 km/s in the air.
The index of refraction of air is 1, the same value as in a vacuum. This refraction index changes when light travels through objects, as it gets slower going through glass, for example. With all of the above then we can infer that light gets slower and its trajectory gets modified slightly as it goes through the eye, and it can be also inferred that every disease that affects the refractive properties of the eye will significantly alter vision.[2][3]
When light waves come across a spherical lens, these waves converge into a focal point, and in the eye, this focal point is projected towards a single area, which is the retina. For this to be accomplished, the crystalline lens must be a dynamic structure. The lens is a capsule filled with water and filamentous proteins; it has a stretched configuration in resting. So, if we imagine this light wave coming across this stretched, very flat lens, then we can assume that light will go farther into the eye because the refraction index is lower. With this crystalline configuration, we can see things clearly even though they are far away because they get projected further into the eye. But when we focus a closer object, then the lens has to change its shape into a more spherical one, in order for the light waves to converge into a closer point, as discussed earlier. This is accomplished by the parasympathetic system. So the nervous system has a role here! Yes, because the parasympathetic system is in charge of constricting fibers in the ciliary muscle. Contraction of these muscle fibers makes the crystalline lens become rounder, as will be discussed later. All of these processes have to be intact for the accommodation reflex to be accomplished.[4]
Cellular Level
At the heart of these organic devices is the visual pigment rhodopsin, a modified molecule of vitamin A. This molecule, which consists of allylic carbons, contains a great deal of conjugated, pi electrons. Recall in organic chemistry an allylic group is a carbon atom singly bonded to another carbon atom, which is in turn double bonded to a carbon atom. The electrons within these alternating pi bonds of the rhodopsin molecule are not as well-defined as the electrons in a saturated carbon chain (no double bonds) or in a singly double-bonded (think simple structure) molecule of ethylene (aka ethene, C2H4).[5]
Rhodopsin consists of the protein scotopsin and the photoreactive chromophore retinal, which is derived from vitamin A. Retinal is covalently bound to one of the protein's lysine residues in a protonated Schiff base (-N+=CH-). The chromophore is the light-absorbing center of the molecule. It functions by facilitating the absorption of photons to potential energy, which supplies the energy needed to allow isomerization of the chromophore molecule from cis to trans (180-degree rotation). Although the reaction mechanism is involved, it can be summarized as:
- Photons elevate electrons within the conjugated pi system to higher energy orbitals (dictated by the level of resonance within the chromophore).
- The molecule rotates around the double-bond, changing from cis to trans configuration.
- By the end of the mechanism, the excited electrons' energy levels have fallen back to the ground state.[6]
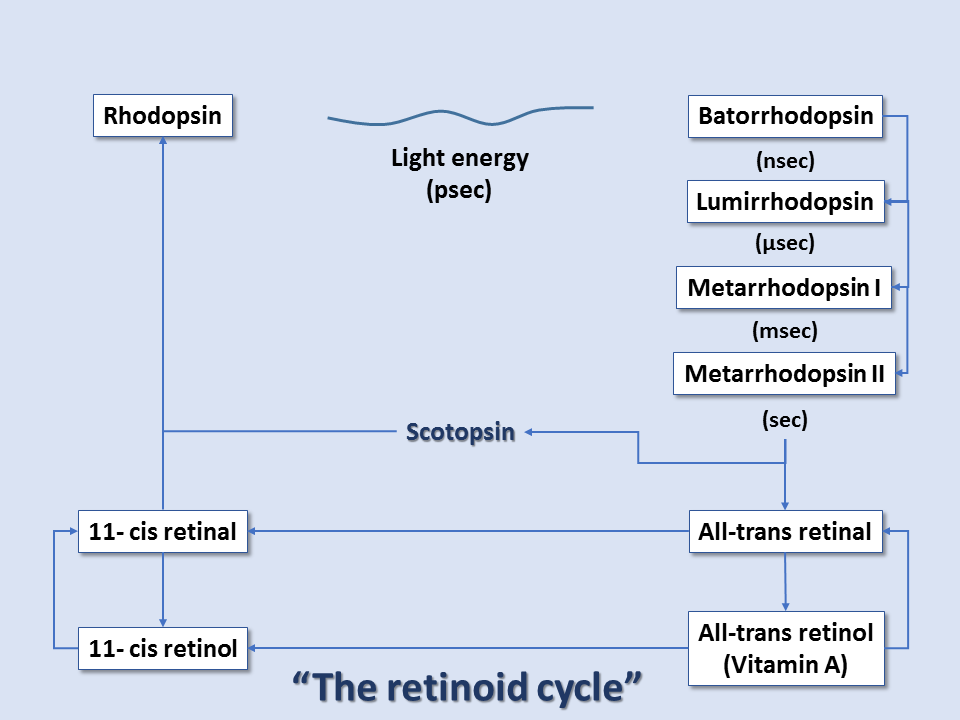
At the reaction completion, the chromophore has changed to the more stable trans configuration. Thus, within rhodopsin, light absorption leads to a chemical reaction that forces part of the rhodopsin molecule to translocate, changing protein conformation, and exposing active sites. This activated form of rhodopsin is known as metarhodopsin II. Before it can get to the metarhodopsin phase, rhodopsin decays through a series of intermediates, and these changes occur in the matter of milliseconds. Metarhodopsin II activates many copies of the G protein transducin (by replacing transducin's GDP with GTP). Many activated transducin complexes activate cyclic nucleotide phosphodiesterase (PDE), which can itself hydrolyze 1000 molecules of cGMP to 5'-GMP per second. cGMP-gated channels in the plasma membrane of these rods (or cones) allow sodium ion influx at high cGMP concentrations; this is balanced by cation exchanger-mediated glutamate efflux, maintaining cell depolarization in dark conditions. At low cGMP concentrations, these channels close, stopping sodium ion influx and reducing glutamate efflux, all leading to cell hyperpolarization in light conditions. Thus, light-induced rod/cone state changes lead to hyperpolarization of the photoreceptor cells. Conversely, photoreceptor cells without the presence of light exist in the depolarized state.[7][8][9]
After this cascade of events, the enzyme rhodopsin kinase quickly binds metarhodopsin II, phosphorylating and halting its activity. The protein arrestin binds phosphorylated metarhodopsin II. Metarhodopsin II is unstable and will split within minutes, leading to opsin and free trans-retinal. Trans-retinal is transported to pigment epithelial cells that convert trans-retinal back to 11-cis-retinal, which eventually is recombined with opsin within cones/rods to reform rhodopsin. Guanylate cyclase restores cGMP concentration, and the cone/rod is ready to respond to another light exposure event.[9]
Additionally, phototransduction is subject to regulation by a calcium-mediated pathway to quickly diffuse a large gradient response, which is important in events such as sudden flashes of light in the dark. In dark conditions, the intracellular calcium level is high due to calcium diffusion through cGMP-gated channels. Lack of frequent light response leads to higher intracellular cGMP concentrations and allows more calcium to enter the cell per second. Calcium ion binding to rhodopsin kinase increases the rate of rhodopsin phosphorylation, reducing transducin activation. Calcium ion binding to guanylate cyclase accelerates the restoration of cGMP concentration. And calcium ion binding to calmodulin increases cGMP affinity to its gated channel.
Development
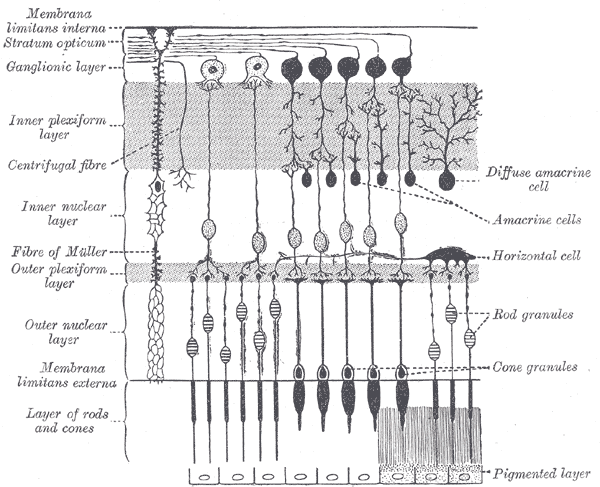
The surface of the human retina contains about three million cones and one hundred million rods, but there are just 1.5 million ganglion cells; meaning that for every ganglion cell, there are sixty rods and two cones. Cones transmit color information, whereas rods have greater sensitivity to low-light conditions. However, the distribution of rods and cones tends to be different depending on the part of the retina. In the central retina, for example, there are almost only cones and a lot of ganglion cells making synapses, which explains why the central retina confers the highest visual acuity. In contrast, in the peripheral retina, there are more rods than cones, and the visual acuity in these peripheral regions is decreased. There are several different types of ganglion cells: W, X, and Y ganglion cells. Some of these are responsible for detecting changes in color intensity (cones), and some are more specialized in detecting changes in contrast (rods). These differences depend on the part of the retina in which the ganglion cells are receiving the stimuli.[10]
The interneural connections of ganglia (bipolar cells) allow for low-level visual processing, adjusting the gain of the signal to transmit light gradients rather than absolute light intensity. Thus, relative differences within the light field and the object’s visual patterns are emphasized, as opposed to binary hit/miss signal information. This process is crucial because rods and cones can distinguish light intensity varying by ten orders of magnitude; however, the ganglia of the optic nerve can only transmit about 1% of this range.[11]
Color vision results from the combination of signals from three pigment types within cones: red, green, and blue pigments that correspond to cone types L, M, and S (RGB-LMS), respectively. Those colors correspond to the wavelengths of peak light absorption intensities of the modified chromophores. Remember, excited electrons are vital to producing Schiff-base modifications, which can be further classified as red shift or blue shift modifications.
Red shift or blue shift modifications denote whether the shift is toward peak absorptions at longer or shorter wavelengths, respectively. The average absorption maxima for 11-cis-retinal occurs at a wavelength of 380nm. If an experimenter were to expose 11-cis-retinal to EM radiation at this wavelength, the 11-cis-retinal would most readily absorb energy, as opposed to with an EM radiation at a wavelength of 280nm. Studies have demonstrated that when retinal is chemically modified to exhibit a more conjugated, distributed pi-electron system, redshift Schiff-base modification is observed. This means the visual pigment exhibits more significant resonance than before, and light is maximally absorbed in a longer wavelength. In contrast, when retinal is chemically modified to exhibit a less conjugated, less distributed pi-electron system, blue shift Schiff-base modification is observed. Here, the visual pigment exhibits less significant resonance than before, and light is maximally absorbed in a shorter wavelength. L cones have peak absorptions at 555-565 nm, M cones at 530-537 nm and S cones at 415-430 nm.[12]
Thus, color vision arises from the shifted cones' peak absorption levels and ultimately the brain's interpretation of the composition of these points of wavelength absorption. The entire pathway is sometimes referred to as the retinoid cycle.
Organ Systems Involved
The sense of vision involves the eye and the series of lenses of which it is composed, the retina, the optic nerve, optic chiasm, the optic tract, the lateral geniculate nuclei in the thalamus and the geniculocalcarine tract that projects to the occipital cortex.
Mechanism
The information coming from the ganglion cells of the retina reaches the optic nerves, and then the action potentials travel to a region called the optic chiasm (where the optic nerve fibers of both eyes cross in the midline and then form the optic tract). The direction of the visual information here is slightly different, as the ipsilateral temporal side of it passes directly into the ipsilateral part of the cortex, whereas the nasal part of vision gets crossed to the contralateral part of the brain, traveling to the opposite occipital cortex. Therefore, downstream to the optic chiasm, every optic tract has information from both eyes, from the temporal ipsilateral part of the visual field and the nasal contralateral part of it. This visual information then gets integrated into the lateral geniculate nuclei of the thalamus and then projects to the visual cortex. Before visual information reaches the thalamus, it can also travel to other structures such as the pretectal nuclei and the superior colliculus in the brainstem (to generate visual reflexes to focus on certain objects) or to the suprachiasmatic nuclei of the hypothalamus (to regulate the circadian rhythms), etc.[13][14]
When the information reaches the thalamus, it has to be ordered like paperwork in an office. So, to accomplish this task, the lateral geniculate nucleus has six layers of neural networks so that information can be integrated and put in order. Layers II, III, and V receive information from the ipsilateral temporal visual field, and layers I, IV and VI receive information from the contralateral nasal visual fields. To make it more interesting, layers I and II are made of magnocellular neurons, and layers III, IV, V, and VI are made of parvocellular neurons. The retina also contains magnocellular and parvocellular neurons, which are subtypes of the ganglion cells (the cells that receive information at the end of the retinal visual pathway). In the retina, the “magnocellular” type of ganglion cells receive information about black and white contrast and rapid changes in object positions, and the “parvocellular” type of neurons receive information about color. So the lateral geniculate nucleus has two layers of neurons dedicated exclusively to the integration of information about black and white contrast and visual field changes, and it has four layers assigned to the combination of color. From here, all of these color and contrast cues go to the visual cortex, where the information is then processed and interpreted.[15]
Related Testing
When evaluating an ophthalmologic complaint, it is very important to delineate the timing of the onset of symptoms. Ocular symptoms may be sudden or progressive, unilateral or bilateral, be associated with pain, photophobia, or discharge. To gain insight from the patient´s medical history, previous ocular conditions, and medications, history of recent ocular surgery or general surgery, the attending clinician must ask intentionally, otherwise, there could be missing information.
A complete physical examination is necessary, inquire about changes in vision with each eye, make a pupillary exam, evaluate extraocular muscle movements and if you are suspecting a CNS lesion or in the optic pathway, test the confrontation visual fields.
Pupil Examination
Pupil abnormalities are one of the most common challenges faced by clinicians. In order to evaluate the pupillary function correctly, one must understand the principles of the physiology of the pupil. The pupil either dilates or constricts. The pupillary dilation is mediated by the sympathetic nervous system, the constricting function is mediated by the parasympathetic nervous system.
- The constricting pupillary pathway starts in the midbrain, there we can find the Edinger - Westphal nucleus. When the eye is exposed to a very near object or to light, this information travels through the optic nerve first, in order for the brain to integrate and process it. Just after the optic chiasm and before information processing at the level of the thalamus (lateral geniculate nuclei), visual information of space and light goes to the EW nucleus after making synapsis in the olivary nuclei. Then, the third cranial nerve acquires parasympathetic fibers originating in these nuclei and travel together. In the subarachnoid space, they travel in the dorsal part of the nerve, very near to the posterior communicating artery. When the oculomotor nerve enters the cavernous sinus, these fibers are configurated more peripherally. Then, the oculomotor nerve reaches the anterior part of the cavernous sinus and soon will reach the superior orbital fissure. Once it reaches the SOF, the oculomotor nerve divides into two divisions: a superior division that innervates the superior palpebrae levator muscle and the superior rectus. The inferior division of the third cranial nerve innervates the rest of the extraocular muscles, except for the lateral rectus (innervated by the abducens) and the superior oblique (innervated by the trochlear nerve). In the posterior aspect of the globe, there is the ciliary ganglion. Here, fibers from the third cranial pair synapse to give origin to the short ciliary nerves that will innervate the pupillary muscles and that will configure the lens alongside the pupil. The near triad is composed of miosis, convergence, and accommodation, this triad is under the influence of more than one brain area, namely the mesencephalic reticular formation, the raphe interpositus, and the superior colliculus.
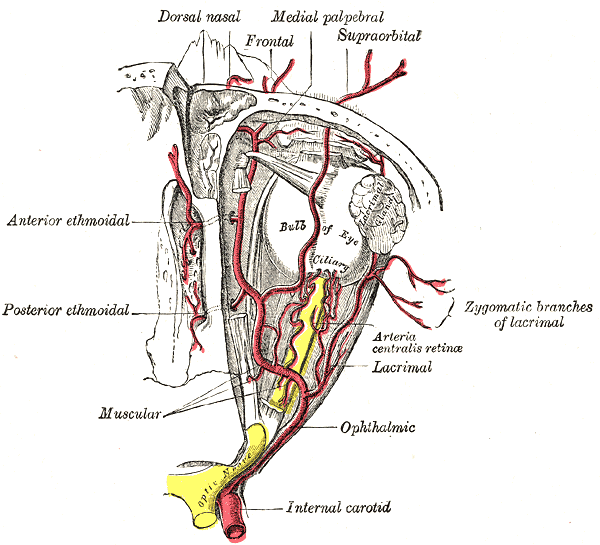
- The dilating pupillary pathway consists of a set of three neurons. The first-order neuron is located in the paraventricular nucleus of the hypothalamus and it travels along the lateral aspect of the brainstem to make synapsis with the second-order neurons located in the ciliospinal nuclei (of Budge), this nucleus extends from C8 to T12 and its fibers run along the lung apex and the subclavian artery and they ascend through the common carotid artery to the third-order neurons that constitute the stellate cervical ganglion. The neurons that originate from this ganglion travel along the internal carotid artery and external carotid artery. The internal carotid artery fibers innervate the Müller muscles and the pupillary dilator muscle; they run along the abducens nerve and the ophthalmic artery. The external carotid fibers innervate the sweat glands of the face.
Anisocoria refers to the discrepancy between the size of both pupils. Physiologic anisocoria is a dilation difference of 1 mm or less between the two pupils. Anisocoria may be monocular or binocular, in miosis or mydriasis.
- Monocular mydriasis: This results from damage to the parasympathetic fibers innervating the pupil. It can be localized to the oculomotor nerve fibers before they reach the ciliary ganglion or after they synapse with it. Always examine the complete function of the third cranial nerve, including the function of the superior and inferior division. Ask yourself if it is a complete or a partial third nerve palsy, is associated with pain, is there any pupil involvement, and if there are signs of abnormal regeneration. The two more common causes of a third nerve palsy are an intracranial aneurysm or an ischemic lesion. Other slow-growing lesions may cause an eye to go mydriatic, like a meningioma. When the ciliary fibers are damaged, they cause a tonic pupil. This means a pupil that stays in mydriasis but typically resolves a little when stimulated by the accommodation reflex. The third cranial nerve is resilient to traumatic injury. When there is a traumatic injury of the oculomotor nerve, there surely is damage to the abducens nerve and trochlear nerve as well. The Adie pupil, as its called, is more common in females, tends to be idiopathic, and may be associated with non-responsive deep tendon reflexes. This is called Holmes-Adie syndrome.
- Binocular mydriasis: These may result from excessive sympathetic stimulation. They may be physiological in the case of an anxiety attack. The concern is the overstimulation of the SNS by drugs, such as cocaine, tricyclic antidepressants, or sympathomimetics. Some cases might be secondary to iris injury or a systemic disease acting as a parasympatholytic such as botulism (never Myasthenia gravis). The bilateral optic neuropathy may present with bilateral mydriasis, as well as diabetic neuropathy.
- Monocular miosis: This presentation arises from the disruption of the sympathetic innervation to the eye. Patients often present with Horner syndrome: is comprised of unilateral miosis, ptosis, and hemifacial ipsilateral anhidrosis. The clinical presentation may vary depending on the order neuron injured. First-order neuron injuries are very uncommon, unless the lesion is at the level of the medulla, being part of a Wallenberg syndrome. Lesion at the level of the ciliospinal nucleus comes from a Brown-Séquard syndrome, commonly. A Pancoast tumor, which is a tumor located a the lung apex, may cause Horner syndrome. Carotid dissection is a rather common cause of Horner syndrome and one should be very aware of this pathology. Note that the syndrome might not be complete in presentation, depending on the level of injury, one might find a partial Horner syndrome. Not every miotic eye is a Horner syndrome. It may be a chronic Adie pupil becoming miotic out of fatigue of the sympathetic nervous system.
- Bilateral miosis: Bilaterally small pupils are not uncommon. This results from a predominance in parasympathetic action over sympathetic action. It may be caused by sedating medications, lesions in the pons, lesions in the diencephalon or chronic reinnervation from a ganglionopathy. One pathology of concern is the Argyll-Robertson pupil. in which both pupils are miotic but are irregular. It is being said that the lesion responsible for this chronic syphilis manifestation is at the level of the midbrain, there's no evidence supporting that theory.[16]
Clinical Significance
Improper Color Vision Recognition/Color Blindness
Many forms of color vision recognition abnormalities are present in the population, with most having a genetic origin. Very few individuals are truly color blind, but instead, individuals who are considered color-blind see a disrupted range of colors. The most common forms are protanopia and deuteranopia, conditions arising from loss of function of one of the cones, leading to dichromic vision. Protanopia is the loss of L cones (red) resulting in green-blue vision only. Deuteranopia is the loss of M cones (green) resulting in red-blue vision only. Both are X-linked alleles, therefore almost exclusively occurring in males, occurring with a prevalence of 1%. Loss of S cones does rarely occur in 0.01% of males and females. In these cases, one of the cones does not function, and one of the others is expressed instead in its place.
Similar to above, but not as severe in its symptoms, is the condition called “anomalous trichromatic vision” (tritanomaly), in which all three cones are present but the color vision is aberrant. The two common forms of color blindness, protanomaly and deuteranomaly, result in loss of L or M cones, respectively, and the lost cones are replaced with cones of intermediate spectral tuning. Both are X-linked and occur in 7% of males.[17]
Diseases affecting color vision but not affecting cones
In addition to disorders of proper color recognition, many diseases in vision display phototransduction defects that affect many portions of the signal pathway and its regulation. Here, not only is color vision function decreased, but the monochromatic vision is worsened as well.
1. Congenital Stationary Night Blindness (CSNB)
One such disease is congenital stationary night blindness. It is a genetic defect resulting in functional cones but dysfunctional rods. In this disease, many potential culprits have been identified including abnormal rhodopsin, arrestin, rod transducin, rod phosphodiesterase, and rhodopsin kinase. Studies have demonstrated that in some populations of this disease, rods are permanently transmitting light signals. In CSNB, b-waves are reduced (in CSNB type 2) or absent (in CSNB type 1) during an electroretinogram (ERG). There are currently no treatments for this disorder.
2. Retinitis Pigmentosa (RP)
Another disease affecting rod function is retinitis pigmentosa, which is a genetically inherited disease characterized by progressive degeneration of the retina, leading to blindness. Frequently, it begins in the early phase as night blindness. Vision loss first occurs in the periphery and progresses towards the center of vision, manifesting as tunnel vision. RP is associated with faulty rod functioning; if cones begin to be affected, then blindness eventually results. RP is characterized by reduced or absent a-waves and b-waves during an ERG. It has a prevalence of 1 in 3500 individuals.
3. Malnutrition-Associated
Deficiency in the essential nutrient vitamin A leads to night blindness, and this can eventually lead to permanent blindness through the deterioration of the receptor outer segments.
Experimental Therapies
Currently, there are no FDA-approved treatments for CSNB. However, the promise of gene-therapy interventions is on the horizon. Recently, FDA-approval was gained for retinal gene therapy (voretigene neparvovec) that utilizes the adeno-associated virus (AAV) and RPE65 gene, which can treat an uncommon form of RP called Leber congenital amaurosis (LCA). This was the first FDA-approved gene therapy for an inherited disorder.[18]