[1]
Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano-Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke-Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S, ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. European heart journal. 2022 Oct 11:43(38):3618-3731. doi: 10.1093/eurheartj/ehac237. Epub
[PubMed PMID: 36017548]
[2]
Condliffe R, Kovacs G. Identifying early pulmonary arterial hypertension in patients with systemic sclerosis. The European respiratory journal. 2018 Apr:51(4):. pii: 1800495. doi: 10.1183/13993003.00495-2018. Epub 2018 Apr 4
[PubMed PMID: 29618608]
[3]
Maron BA, Brittain EL, Choudhary G, Gladwin MT. Redefining pulmonary hypertension. The Lancet. Respiratory medicine. 2018 Mar:6(3):168-170. doi: 10.1016/S2213-2600(17)30498-8. Epub 2017 Dec 18
[PubMed PMID: 29269004]
[4]
Maron BA, Wertheim BM, Gladwin MT. Under Pressure to Clarify Pulmonary Hypertension Clinical Risk. American journal of respiratory and critical care medicine. 2018 Feb 15:197(4):423-426. doi: 10.1164/rccm.201711-2306ED. Epub
[PubMed PMID: 29216444]
[5]
Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. The European respiratory journal. 2009 Oct:34(4):888-94. doi: 10.1183/09031936.00145608. Epub 2009 Mar 26
[PubMed PMID: 19324955]
Level 1 (high-level) evidence
[6]
Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. The European respiratory journal. 2019 Jan:53(1):. doi: 10.1183/13993003.01913-2018. Epub 2019 Jan 24
[PubMed PMID: 30545968]
[7]
Bentley RF, Barker M, Esfandiari S, Wright SP, Valle FH, Granton JT, Mak S. Normal and Abnormal Relationships of Pulmonary Artery to Wedge Pressure During Exercise. Journal of the American Heart Association. 2020 Nov 17:9(22):e016339. doi: 10.1161/JAHA.120.016339. Epub 2020 Nov 6
[PubMed PMID: 33153377]
[8]
Eisman AS, Shah RV, Dhakal BP, Pappagianopoulos PP, Wooster L, Bailey C, Cunningham TF, Hardin KM, Baggish AL, Ho JE, Malhotra R, Lewis GD. Pulmonary Capillary Wedge Pressure Patterns During Exercise Predict Exercise Capacity and Incident Heart Failure. Circulation. Heart failure. 2018 May:11(5):e004750. doi: 10.1161/CIRCHEARTFAILURE.117.004750. Epub
[PubMed PMID: 29695381]
[9]
Garcia-Rivas G, Jerjes-Sánchez C, Rodriguez D, Garcia-Pelaez J, Trevino V. A systematic review of genetic mutations in pulmonary arterial hypertension. BMC medical genetics. 2017 Aug 2:18(1):82. doi: 10.1186/s12881-017-0440-5. Epub 2017 Aug 2
[PubMed PMID: 28768485]
Level 1 (high-level) evidence
[10]
Simonneau G, Galiè N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, Lebrec D, Speich R, Beghetti M, Rich S, Fishman A. Clinical classification of pulmonary hypertension. Journal of the American College of Cardiology. 2004 Jun 16:43(12 Suppl S):5S-12S
[PubMed PMID: 15194173]
[11]
Hoeper MM, Humbert M, Souza R, Idrees M, Kawut SM, Sliwa-Hahnle K, Jing ZC, Gibbs JS. A global view of pulmonary hypertension. The Lancet. Respiratory medicine. 2016 Apr:4(4):306-22. doi: 10.1016/S2213-2600(15)00543-3. Epub 2016 Mar 12
[PubMed PMID: 26975810]
[12]
Leber L, Beaudet A, Muller A. Epidemiology of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: identification of the most accurate estimates from a systematic literature review. Pulmonary circulation. 2021 Jan-Mar:11(1):2045894020977300. doi: 10.1177/2045894020977300. Epub 2021 Jan 7
[PubMed PMID: 33456755]
Level 1 (high-level) evidence
[13]
Montani D, Girerd B, Jaïs X, Laveneziana P, Lau EMT, Bouchachi A, Hascoët S, Günther S, Godinas L, Parent F, Guignabert C, Beurnier A, Chemla D, Hervé P, Eyries M, Soubrier F, Simonneau G, Sitbon O, Savale L, Humbert M. Screening for pulmonary arterial hypertension in adults carrying a BMPR2 mutation. The European respiratory journal. 2021 Jul:58(1):. doi: 10.1183/13993003.04229-2020. Epub 2021 Jul 22
[PubMed PMID: 33380512]
[14]
Lau EMT, Giannoulatou E, Celermajer DS, Humbert M. Epidemiology and treatment of pulmonary arterial hypertension. Nature reviews. Cardiology. 2017 Oct:14(10):603-614. doi: 10.1038/nrcardio.2017.84. Epub 2017 Jun 8
[PubMed PMID: 28593996]
[15]
Rosenkranz S, Gibbs JS, Wachter R, De Marco T, Vonk-Noordegraaf A, Vachiéry JL. Left ventricular heart failure and pulmonary hypertension. European heart journal. 2016 Mar 21:37(12):942-54. doi: 10.1093/eurheartj/ehv512. Epub 2015 Oct 27
[PubMed PMID: 26508169]
[16]
Lam CS, Roger VL, Rodeheffer RJ, Borlaug BA, Enders FT, Redfield MM. Pulmonary hypertension in heart failure with preserved ejection fraction: a community-based study. Journal of the American College of Cardiology. 2009 Mar 31:53(13):1119-26. doi: 10.1016/j.jacc.2008.11.051. Epub
[PubMed PMID: 19324256]
[17]
Tichelbäcker T, Dumitrescu D, Gerhardt F, Stern D, Wissmüller M, Adam M, Schmidt T, Frerker C, Pfister R, Halbach M, Baldus S, Rosenkranz S. Pulmonary hypertension and valvular heart disease. Herz. 2019 Sep:44(6):491-501. doi: 10.1007/s00059-019-4823-6. Epub
[PubMed PMID: 31312873]
[18]
Weber L, Rickli H, Haager PK, Joerg L, Weilenmann D, Brenner R, Taramasso M, Baier P, Maisano F, Maeder MT. Haemodynamic mechanisms and long-term prognostic impact of pulmonary hypertension in patients with severe aortic stenosis undergoing valve replacement. European journal of heart failure. 2019 Feb:21(2):172-181. doi: 10.1002/ejhf.1322. Epub 2018 Oct 17
[PubMed PMID: 30328215]
[19]
Nathan SD, Barbera JA, Gaine SP, Harari S, Martinez FJ, Olschewski H, Olsson KM, Peacock AJ, Pepke-Zaba J, Provencher S, Weissmann N, Seeger W. Pulmonary hypertension in chronic lung disease and hypoxia. The European respiratory journal. 2019 Jan:53(1):. doi: 10.1183/13993003.01914-2018. Epub 2019 Jan 24
[PubMed PMID: 30545980]
[20]
Hurdman J, Condliffe R, Elliot CA, Swift A, Rajaram S, Davies C, Hill C, Hamilton N, Armstrong IJ, Billings C, Pollard L, Wild JM, Lawrie A, Lawson R, Sabroe I, Kiely DG. Pulmonary hypertension in COPD: results from the ASPIRE registry. The European respiratory journal. 2013 Jun:41(6):1292-301. doi: 10.1183/09031936.00079512. Epub 2012 Sep 27
[PubMed PMID: 23018917]
[21]
Delcroix M, Torbicki A, Gopalan D, Sitbon O, Klok FA, Lang I, Jenkins D, Kim NH, Humbert M, Jais X, Vonk Noordegraaf A, Pepke-Zaba J, Brénot P, Dorfmuller P, Fadel E, Ghofrani HA, Hoeper MM, Jansa P, Madani M, Matsubara H, Ogo T, Grünig E, D'Armini A, Galie N, Meyer B, Corkery P, Meszaros G, Mayer E, Simonneau G. ERS statement on chronic thromboembolic pulmonary hypertension. The European respiratory journal. 2021 Jun:57(6):. pii: 2002828. doi: 10.1183/13993003.02828-2020. Epub 2021 Jun 17
[PubMed PMID: 33334946]
[22]
Kramm T, Wilkens H, Fuge J, Schäfers HJ, Guth S, Wiedenroth CB, Weingard B, Huscher D, Pittrow D, Cebotari S, Hoeper MM, Mayer E, Olsson KM. Incidence and characteristics of chronic thromboembolic pulmonary hypertension in Germany. Clinical research in cardiology : official journal of the German Cardiac Society. 2018 Jul:107(7):548-553. doi: 10.1007/s00392-018-1215-5. Epub 2018 Feb 15
[PubMed PMID: 29450722]
[23]
Shlobin OA, Kouranos V, Barnett SD, Alhamad EH, Culver DA, Barney J, Cordova FC, Carmona EM, Scholand MB, Wijsenbeek M, Ganesh S, Lower EE, Engel PJ, Wort J, Price L, Wells AU, Nathan SD, Baughman RP. Physiological predictors of survival in patients with sarcoidosis-associated pulmonary hypertension: results from an international registry. The European respiratory journal. 2020 May:55(5):. pii: 1901747. doi: 10.1183/13993003.01747-2019. Epub 2020 May 14
[PubMed PMID: 32139456]
[24]
Boucly A, Cottin V, Nunes H, Jaïs X, Tazi A, Prévôt G, Reynaud-Gaubert M, Dromer C, Viacroze C, Horeau-Langlard D, Pison C, Bergot E, Traclet J, Weatherald J, Simonneau G, Valeyre D, Montani D, Humbert M, Sitbon O, Savale L. Management and long-term outcomes of sarcoidosis-associated pulmonary hypertension. The European respiratory journal. 2017 Oct:50(4):. pii: 1700465. doi: 10.1183/13993003.00465-2017. Epub 2017 Oct 19
[PubMed PMID: 29051269]
[25]
Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). The European respiratory journal. 2015 Oct:46(4):903-75. doi: 10.1183/13993003.01032-2015. Epub 2015 Aug 29
[PubMed PMID: 26318161]
[26]
Armstrong I, Billings C, Kiely DG, Yorke J, Harries C, Clayton S, Gin-Sing W. The patient experience of pulmonary hypertension: a large cross-sectional study of UK patients. BMC pulmonary medicine. 2019 Mar 21:19(1):67. doi: 10.1186/s12890-019-0827-5. Epub 2019 Mar 21
[PubMed PMID: 30898139]
Level 2 (mid-level) evidence
[27]
Strange G, Gabbay E, Kermeen F, Williams T, Carrington M, Stewart S, Keogh A. Time from symptoms to definitive diagnosis of idiopathic pulmonary arterial hypertension: The delay study. Pulmonary circulation. 2013 Jan:3(1):89-94. doi: 10.4103/2045-8932.109919. Epub
[PubMed PMID: 23662179]
[28]
Rich JD, Thenappan T, Freed B, Patel AR, Thisted RA, Childers R, Archer SL. QTc prolongation is associated with impaired right ventricular function and predicts mortality in pulmonary hypertension. International journal of cardiology. 2013 Aug 10:167(3):669-76. doi: 10.1016/j.ijcard.2012.03.071. Epub 2012 Mar 27
[PubMed PMID: 22459397]
[29]
Sun PY, Jiang X, Gomberg-Maitland M, Zhao QH, He J, Yuan P, Zhang R, Jing ZC. Prolonged QRS duration: a new predictor of adverse outcome in idiopathic pulmonary arterial hypertension. Chest. 2012 Feb:141(2):374-380. doi: 10.1378/chest.10-3331. Epub 2011 Jul 21
[PubMed PMID: 21778258]
[30]
Henkens IR, Gan CT, van Wolferen SA, Hew M, Boonstra A, Twisk JWR, Kamp O, van der Wall EE, Schalij MJ, Vonk Noordegraaf A, Vliegen HW. ECG monitoring of treatment response in pulmonary arterial hypertension patients. Chest. 2008 Dec:134(6):1250-1257. doi: 10.1378/chest.08-0461. Epub 2008 Jul 18
[PubMed PMID: 18641107]
[31]
Bossone E, Paciocco G, Iarussi D, Agretto A, Iacono A, Gillespie BW, Rubenfire M. The prognostic role of the ECG in primary pulmonary hypertension. Chest. 2002 Feb:121(2):513-8
[PubMed PMID: 11834666]
[32]
Wanamaker B, Cascino T, McLaughlin V, Oral H, Latchamsetty R, Siontis KC. Atrial Arrhythmias in Pulmonary Hypertension: Pathogenesis, Prognosis and Management. Arrhythmia & electrophysiology review. 2018 Mar:7(1):43-48. doi: 10.15420/aer.2018.3.2. Epub
[PubMed PMID: 29636972]
[33]
Ascha M, Renapurkar RD, Tonelli AR. A review of imaging modalities in pulmonary hypertension. Annals of thoracic medicine. 2017 Apr-Jun:12(2):61-73. doi: 10.4103/1817-1737.203742. Epub
[PubMed PMID: 28469715]
[34]
Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, Langleben D, Manes A, Satoh T, Torres F, Wilkins MR, Badesch DB. Definitions and diagnosis of pulmonary hypertension. Journal of the American College of Cardiology. 2013 Dec 24:62(25 Suppl):D42-50. doi: 10.1016/j.jacc.2013.10.032. Epub
[PubMed PMID: 24355641]
[35]
Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Koerner SK. Primary pulmonary hypertension. A national prospective study. Annals of internal medicine. 1987 Aug:107(2):216-23
[PubMed PMID: 3605900]
[36]
Trip P, Nossent EJ, de Man FS, van den Berk IA, Boonstra A, Groepenhoff H, Leter EM, Westerhof N, Grünberg K, Bogaard HJ, Vonk-Noordegraaf A. Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: patient characteristics and treatment responses. The European respiratory journal. 2013 Dec:42(6):1575-85. doi: 10.1183/09031936.00184412. Epub 2013 Aug 15
[PubMed PMID: 23949959]
[37]
Sun XG, Hansen JE, Oudiz RJ, Wasserman K. Pulmonary function in primary pulmonary hypertension. Journal of the American College of Cardiology. 2003 Mar 19:41(6):1028-35
[PubMed PMID: 12651053]
[38]
Mélot C, Naeije R. Pulmonary vascular diseases. Comprehensive Physiology. 2011 Apr:1(2):593-619. doi: 10.1002/cphy.c090014. Epub
[PubMed PMID: 23737196]
[39]
Harbaum L, Fuge J, Kamp JC, Hennigs JK, Simon M, Sinning C, Oqueka T, Grimminger J, Olsson KM, Hoeper MM, Klose H. Blood carbon dioxide tension and risk in pulmonary arterial hypertension. International journal of cardiology. 2020 Nov 1:318():131-137. doi: 10.1016/j.ijcard.2020.06.069. Epub 2020 Jul 4
[PubMed PMID: 32634498]
[40]
Jilwan FN, Escourrou P, Garcia G, Jaïs X, Humbert M, Roisman G. High occurrence of hypoxemic sleep respiratory disorders in precapillary pulmonary hypertension and mechanisms. Chest. 2013 Jan:143(1):47-55. doi: 10.1378/chest.11-3124. Epub
[PubMed PMID: 22878784]
[41]
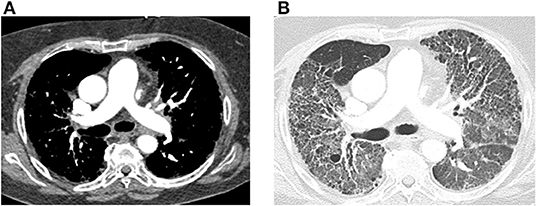
Swift AJ, Dwivedi K, Johns C, Garg P, Chin M, Currie BJ, Rothman AM, Capener D, Shahin Y, Elliot CA, Charalampopolous T, Sabroe I, Rajaram S, Hill C, Wild JM, Condliffe R, Kiely DG. Diagnostic accuracy of CT pulmonary angiography in suspected pulmonary hypertension. European radiology. 2020 Sep:30(9):4918-4929. doi: 10.1007/s00330-020-06846-1. Epub 2020 Apr 27
[PubMed PMID: 32342182]
[42]
Dong C, Zhou M, Liu D, Long X, Guo T, Kong X. Diagnostic accuracy of computed tomography for chronic thromboembolic pulmonary hypertension: a systematic review and meta-analysis. PloS one. 2015:10(4):e0126985. doi: 10.1371/journal.pone.0126985. Epub 2015 Apr 29
[PubMed PMID: 25923810]
Level 1 (high-level) evidence
[43]
Rajaram S, Swift AJ, Capener D, Telfer A, Davies C, Hill C, Condliffe R, Elliot C, Hurdman J, Kiely DG, Wild JM. Diagnostic accuracy of contrast-enhanced MR angiography and unenhanced proton MR imaging compared with CT pulmonary angiography in chronic thromboembolic pulmonary hypertension. European radiology. 2012 Feb:22(2):310-7. doi: 10.1007/s00330-011-2252-x. Epub 2011 Sep 2
[PubMed PMID: 21887483]
[44]
Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M, ESC Scientific Document Group. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). European heart journal. 2016 Jan 1:37(1):67-119. doi: 10.1093/eurheartj/ehv317. Epub 2015 Aug 29
[PubMed PMID: 26320113]
[45]
Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski L, Spencer KT, Tsang W, Voigt JU. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Journal of the American Society of Echocardiography : official publication of the American Society of Echocardiography. 2015 Jan:28(1):1-39.e14. doi: 10.1016/j.echo.2014.10.003. Epub
[PubMed PMID: 25559473]
[46]
Rudski LG, Lai WW, Afilalo J, Hua L, Handschumacher MD, Chandrasekaran K, Solomon SD, Louie EK, Schiller NB. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. Journal of the American Society of Echocardiography : official publication of the American Society of Echocardiography. 2010 Jul:23(7):685-713; quiz 786-8. doi: 10.1016/j.echo.2010.05.010. Epub
[PubMed PMID: 20620859]
[47]
Foale R, Nihoyannopoulos P, McKenna W, Kleinebenne A, Nadazdin A, Rowland E, Smith G. Echocardiographic measurement of the normal adult right ventricle. British heart journal. 1986 Jul:56(1):33-44
[PubMed PMID: 3730205]
[48]
Sun XG, Hansen JE, Oudiz RJ, Wasserman K. Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation. 2001 Jul 24:104(4):429-35
[PubMed PMID: 11468205]
[49]
Dumitrescu D, Nagel C, Kovacs G, Bollmann T, Halank M, Winkler J, Hellmich M, Grünig E, Olschewski H, Ewert R, Rosenkranz S. Cardiopulmonary exercise testing for detecting pulmonary arterial hypertension in systemic sclerosis. Heart (British Cardiac Society). 2017 May:103(10):774-782. doi: 10.1136/heartjnl-2016-309981. Epub 2017 Jan 6
[PubMed PMID: 28062514]
[50]
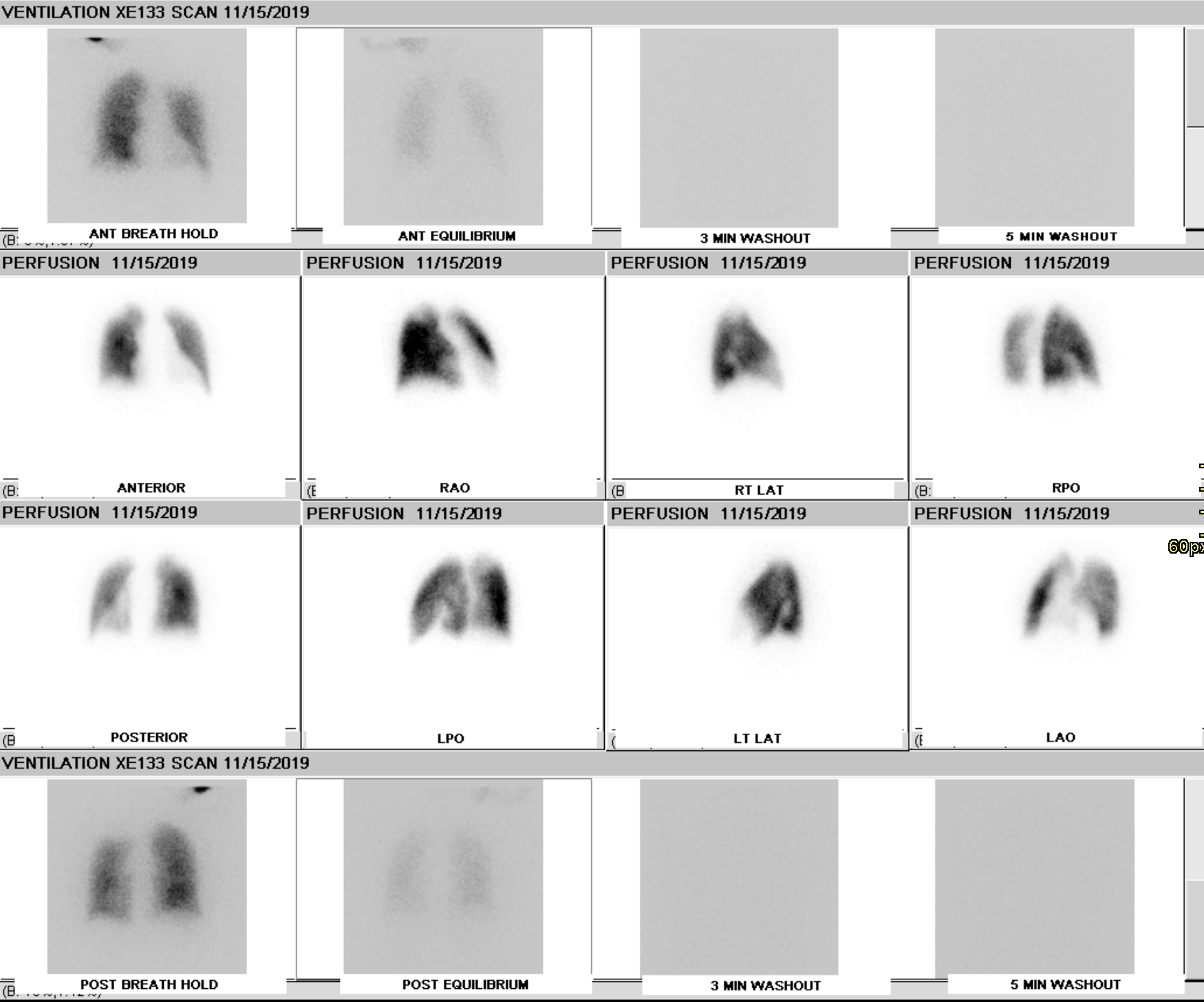
He J, Fang W, Lv B, He JG, Xiong CM, Liu ZH, He ZX. Diagnosis of chronic thromboembolic pulmonary hypertension: comparison of ventilation/perfusion scanning and multidetector computed tomography pulmonary angiography with pulmonary angiography. Nuclear medicine communications. 2012 May:33(5):459-63. doi: 10.1097/MNM.0b013e32835085d9. Epub
[PubMed PMID: 22262242]
[51]
Tunariu N, Gibbs SJ, Win Z, Gin-Sing W, Graham A, Gishen P, Al-Nahhas A. Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboembolic pulmonary disease as a treatable cause of pulmonary hypertension. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2007 May:48(5):680-4
[PubMed PMID: 17475953]
[52]
Johns CS, Swift AJ, Rajaram S, Hughes PJC, Capener DJ, Kiely DG, Wild JM. Lung perfusion: MRI vs. SPECT for screening in suspected chronic thromboembolic pulmonary hypertension. Journal of magnetic resonance imaging : JMRI. 2017 Dec:46(6):1693-1697. doi: 10.1002/jmri.25714. Epub 2017 Apr 4
[PubMed PMID: 28376242]
[53]
Meng JJ, Zhang LJ, Wang Q, Fang W, Dai HJ, Yan J, Wang T, Yao ZM, He J, Li M, Mi HZ, Jiao J, Zheng YM. [A comparison of ventilation/perfusion single photon emission CT and CT pulmonary angiography for diagnosis of pulmonary embolism]. Zhonghua jie he he hu xi za zhi = Zhonghua jiehe he huxi zazhi = Chinese journal of tuberculosis and respiratory diseases. 2013 Mar:36(3):177-81
[PubMed PMID: 23856139]
[54]
Rajaram S, Swift AJ, Telfer A, Hurdman J, Marshall H, Lorenz E, Capener D, Davies C, Hill C, Elliot C, Condliffe R, Wild JM, Kiely DG. 3D contrast-enhanced lung perfusion MRI is an effective screening tool for chronic thromboembolic pulmonary hypertension: results from the ASPIRE Registry. Thorax. 2013 Jul:68(7):677-8. doi: 10.1136/thoraxjnl-2012-203020. Epub 2013 Jan 24
[PubMed PMID: 23349220]
[55]
Swift AJ, Lu H, Uthoff J, Garg P, Cogliano M, Taylor J, Metherall P, Zhou S, Johns CS, Alabed S, Condliffe RA, Lawrie A, Wild JM, Kiely DG. A machine learning cardiac magnetic resonance approach to extract disease features and automate pulmonary arterial hypertension diagnosis. European heart journal. Cardiovascular Imaging. 2021 Jan 22:22(2):236-245. doi: 10.1093/ehjci/jeaa001. Epub
[PubMed PMID: 31998956]
[56]
Rosenkranz S, Howard LS, Gomberg-Maitland M, Hoeper MM. Systemic Consequences of Pulmonary Hypertension and Right-Sided Heart Failure. Circulation. 2020 Feb 25:141(8):678-693. doi: 10.1161/CIRCULATIONAHA.116.022362. Epub 2020 Feb 24
[PubMed PMID: 32091921]
[57]
Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, Soubrier F, Trembath RC, Loyd JE. Genetics and genomics of pulmonary arterial hypertension. The European respiratory journal. 2019 Jan:53(1):. doi: 10.1183/13993003.01899-2018. Epub 2019 Jan 24
[PubMed PMID: 30545973]
[58]
Song J, Eichstaedt CA, Viales RR, Benjamin N, Harutyunova S, Fischer C, Grünig E, Hinderhofer K. Identification of genetic defects in pulmonary arterial hypertension by a new gene panel diagnostic tool. Clinical science (London, England : 1979). 2016 Nov 1:130(22):2043-2052. doi: 10.1042/CS20160531. Epub 2016 Sep 9
[PubMed PMID: 27613157]
[59]
Hoeper MM, Lee SH, Voswinckel R, Palazzini M, Jais X, Marinelli A, Barst RJ, Ghofrani HA, Jing ZC, Opitz C, Seyfarth HJ, Halank M, McLaughlin V, Oudiz RJ, Ewert R, Wilkens H, Kluge S, Bremer HC, Baroke E, Rubin LJ. Complications of right heart catheterization procedures in patients with pulmonary hypertension in experienced centers. Journal of the American College of Cardiology. 2006 Dec 19:48(12):2546-52
[PubMed PMID: 17174196]
[60]
Kovacs G, Avian A, Pienn M, Naeije R, Olschewski H. Reading pulmonary vascular pressure tracings. How to handle the problems of zero leveling and respiratory swings. American journal of respiratory and critical care medicine. 2014 Aug 1:190(3):252-7. doi: 10.1164/rccm.201402-0269PP. Epub
[PubMed PMID: 24869464]
[61]
Hoeper MM, Maier R, Tongers J, Niedermeyer J, Hohlfeld JM, Hamm M, Fabel H. Determination of cardiac output by the Fick method, thermodilution, and acetylene rebreathing in pulmonary hypertension. American journal of respiratory and critical care medicine. 1999 Aug:160(2):535-41
[PubMed PMID: 10430725]
[62]
Robbins IM, Hemnes AR, Pugh ME, Brittain EL, Zhao DX, Piana RN, Fong PP, Newman JH. High prevalence of occult pulmonary venous hypertension revealed by fluid challenge in pulmonary hypertension. Circulation. Heart failure. 2014 Jan:7(1):116-22. doi: 10.1161/CIRCHEARTFAILURE.113.000468. Epub 2013 Dec 2
[PubMed PMID: 24297689]
[63]
Fox BD, Shimony A, Langleben D, Hirsch A, Rudski L, Schlesinger R, Eisenberg MJ, Joyal D, Hudson M, Boutet K, Serban A, Masetto A, Baron M. High prevalence of occult left heart disease in scleroderma-pulmonary hypertension. The European respiratory journal. 2013 Oct:42(4):1083-91. doi: 10.1183/09031936.00091212. Epub 2012 Dec 20
[PubMed PMID: 23258775]
[64]
Hoeper MM, Pittrow D, Opitz C, Gibbs JSR, Rosenkranz S, Grünig E, Olsson KM, Huscher D. Risk assessment in pulmonary arterial hypertension. The European respiratory journal. 2018 Mar:51(3):. pii: 1702606. doi: 10.1183/13993003.02606-2017. Epub 2018 Mar 29
[PubMed PMID: 29599117]
[65]
Boucly A, Weatherald J, Savale L, Jaïs X, Cottin V, Prevot G, Picard F, de Groote P, Jevnikar M, Bergot E, Chaouat A, Chabanne C, Bourdin A, Parent F, Montani D, Simonneau G, Humbert M, Sitbon O. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. The European respiratory journal. 2017 Aug:50(2):. pii: 1700889. doi: 10.1183/13993003.00889-2017. Epub 2017 Aug 3
[PubMed PMID: 28775050]
[66]
Hoeper MM, Kramer T, Pan Z, Eichstaedt CA, Spiesshoefer J, Benjamin N, Olsson KM, Meyer K, Vizza CD, Vonk-Noordegraaf A, Distler O, Opitz C, Gibbs JSR, Delcroix M, Ghofrani HA, Huscher D, Pittrow D, Rosenkranz S, Grünig E. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. The European respiratory journal. 2017 Aug:50(2):. pii: 1700740. doi: 10.1183/13993003.00740-2017. Epub 2017 Aug 3
[PubMed PMID: 28775047]
[67]
Kylhammar D, Kjellström B, Hjalmarsson C, Jansson K, Nisell M, Söderberg S, Wikström G, Rådegran G. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. European heart journal. 2018 Dec 14:39(47):4175-4181. doi: 10.1093/eurheartj/ehx257. Epub
[PubMed PMID: 28575277]
[68]
Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaïci A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M, Hachulla E, Cottin V, Degano B, Jaïs X, Montani D, Souza R, Simonneau G. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010 Jul 13:122(2):156-63. doi: 10.1161/CIRCULATIONAHA.109.911818. Epub 2010 Jun 28
[PubMed PMID: 20585011]
[69]
Humbert M, Sitbon O, Yaïci A, Montani D, O'Callaghan DS, Jaïs X, Parent F, Savale L, Natali D, Günther S, Chaouat A, Chabot F, Cordier JF, Habib G, Gressin V, Jing ZC, Souza R, Simonneau G, French Pulmonary Arterial Hypertension Network. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. The European respiratory journal. 2010 Sep:36(3):549-55. doi: 10.1183/09031936.00057010. Epub 2010 Jun 18
[PubMed PMID: 20562126]
[70]
Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010 Jul 13:122(2):164-72. doi: 10.1161/CIRCULATIONAHA.109.898122. Epub 2010 Jun 28
[PubMed PMID: 20585012]
[71]
Benza RL, Gomberg-Maitland M, Miller DP, Frost A, Frantz RP, Foreman AJ, Badesch DB, McGoon MD. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest. 2012 Feb:141(2):354-362. doi: 10.1378/chest.11-0676. Epub 2011 Jun 16
[PubMed PMID: 21680644]
[72]
Benza RL, Miller DP, Foreman AJ, Frost AE, Badesch DB, Benton WW, McGoon MD. Prognostic implications of serial risk score assessments in patients with pulmonary arterial hypertension: a Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) analysis. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2015 Mar:34(3):356-61. doi: 10.1016/j.healun.2014.09.016. Epub 2014 Sep 28
[PubMed PMID: 25447572]
[73]
Chakinala MM, Coyne DW, Benza RL, Frost AE, McGoon MD, Hartline BK, Frantz RP, Selej M, Zhao C, Mink DR, Farber HW. Impact of declining renal function on outcomes in pulmonary arterial hypertension: A REVEAL registry analysis. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2018 Jun:37(6):696-705. doi: 10.1016/j.healun.2017.10.028. Epub 2017 Nov 6
[PubMed PMID: 29174533]
[74]
Frost AE, Badesch DB, Miller DP, Benza RL, Meltzer LA, McGoon MD. Evaluation of the predictive value of a clinical worsening definition using 2-year outcomes in patients with pulmonary arterial hypertension: a REVEAL Registry analysis. Chest. 2013 Nov:144(5):1521-1529. doi: 10.1378/chest.12-3023. Epub
[PubMed PMID: 23907471]
[75]
Benza RL, Kanwar MK, Raina A, Scott JV, Zhao CL, Selej M, Elliott CG, Farber HW. Development and Validation of an Abridged Version of the REVEAL 2.0 Risk Score Calculator, REVEAL Lite 2, for Use in Patients With Pulmonary Arterial Hypertension. Chest. 2021 Jan:159(1):337-346. doi: 10.1016/j.chest.2020.08.2069. Epub 2020 Sep 1
[PubMed PMID: 32882243]
Level 1 (high-level) evidence
[76]
Weatherald J, Sitbon O, Humbert M. Validation of a risk assessment instrument for pulmonary arterial hypertension. European heart journal. 2018 Dec 14:39(47):4182-4185. doi: 10.1093/eurheartj/ehx301. Epub
[PubMed PMID: 28637288]
Level 1 (high-level) evidence
[77]
Sitbon O, Humbert M, Jaïs X, Ioos V, Hamid AM, Provencher S, Garcia G, Parent F, Hervé P, Simonneau G. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005 Jun 14:111(23):3105-11
[PubMed PMID: 15939821]
[78]
Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. The New England journal of medicine. 1992 Jul 9:327(2):76-81
[PubMed PMID: 1603139]
[79]
Clozel M, Maresta A, Humbert M. Endothelin receptor antagonists. Handbook of experimental pharmacology. 2013:218():199-227. doi: 10.1007/978-3-642-38664-0_9. Epub
[PubMed PMID: 24092342]
[80]
Wharton J, Strange JW, Møller GM, Growcott EJ, Ren X, Franklyn AP, Phillips SC, Wilkins MR. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. American journal of respiratory and critical care medicine. 2005 Jul 1:172(1):105-13
[PubMed PMID: 15817798]
[81]
Tantini B, Manes A, Fiumana E, Pignatti C, Guarnieri C, Zannoli R, Branzi A, Galié N. Antiproliferative effect of sildenafil on human pulmonary artery smooth muscle cells. Basic research in cardiology. 2005 Mar:100(2):131-8
[PubMed PMID: 15739122]
[82]
Ghofrani HA, Voswinckel R, Reichenberger F, Olschewski H, Haredza P, Karadaş B, Schermuly RT, Weissmann N, Seeger W, Grimminger F. Differences in hemodynamic and oxygenation responses to three different phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension: a randomized prospective study. Journal of the American College of Cardiology. 2004 Oct 6:44(7):1488-96
[PubMed PMID: 15464333]
Level 1 (high-level) evidence
[83]
Jones DA, Benjamin CW, Linseman DA. Activation of thromboxane and prostacyclin receptors elicits opposing effects on vascular smooth muscle cell growth and mitogen-activated protein kinase signaling cascades. Molecular pharmacology. 1995 Nov:48(5):890-6
[PubMed PMID: 7476920]
[84]
Doran AK, Ivy DD, Barst RJ, Hill N, Murali S, Benza RL, Scientific Leadership Council of the Pulmonary Hypertension Association. Guidelines for the prevention of central venous catheter-related blood stream infections with prostanoid therapy for pulmonary arterial hypertension. International journal of clinical practice. Supplement. 2008 Jul:(160):5-9. doi: 10.1111/j.1742-1241.2008.01811.x. Epub
[PubMed PMID: 18638170]
[85]
Simonneau G, Barst RJ, Galie N, Naeije R, Rich S, Bourge RC, Keogh A, Oudiz R, Frost A, Blackburn SD, Crow JW, Rubin LJ, Treprostinil Study Group. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. American journal of respiratory and critical care medicine. 2002 Mar 15:165(6):800-4
[PubMed PMID: 11897647]
Level 1 (high-level) evidence
[86]
Khan MS, Memon MM, Amin E, Yamani N, Khan SU, Figueredo VM, Deo S, Rich JD, Benza RL, Krasuski RA. Use of Balloon Atrial Septostomy in Patients With Advanced Pulmonary Arterial Hypertension: A Systematic Review and Meta-Analysis. Chest. 2019 Jul:156(1):53-63. doi: 10.1016/j.chest.2019.03.003. Epub 2019 Mar 23
[PubMed PMID: 30910639]
Level 1 (high-level) evidence
[87]
Sandoval J, Gaspar J, Pulido T, Bautista E, Martínez-Guerra ML, Zeballos M, Palomar A, Gómez A. Graded balloon dilation atrial septostomy in severe primary pulmonary hypertension. A therapeutic alternative for patients nonresponsive to vasodilator treatment. Journal of the American College of Cardiology. 1998 Aug:32(2):297-304
[PubMed PMID: 9708453]
[88]
Rothman AMK, Vachiery JL, Howard LS, Mikhail GW, Lang IM, Jonas M, Kiely DG, Shav D, Shabtay O, Avriel A, Lewis GD, Rosenzweig EB, Kirtane AJ, Kim NH, Mahmud E, McLaughlain VV, Chetcuti S, Leon MB, Ben-Yehuda O, Rubin LJ. Intravascular Ultrasound Pulmonary Artery Denervation to Treat Pulmonary Arterial Hypertension (TROPHY1): Multicenter, Early Feasibility Study. JACC. Cardiovascular interventions. 2020 Apr 27:13(8):989-999. doi: 10.1016/j.jcin.2019.12.027. Epub
[PubMed PMID: 32327095]
Level 2 (mid-level) evidence
[89]
Chen SL, Zhang FF, Xu J, Xie DJ, Zhou L, Nguyen T, Stone GW. Pulmonary artery denervation to treat pulmonary arterial hypertension: the single-center, prospective, first-in-man PADN-1 study (first-in-man pulmonary artery denervation for treatment of pulmonary artery hypertension). Journal of the American College of Cardiology. 2013 Sep 17:62(12):1092-1100. doi: 10.1016/j.jacc.2013.05.075. Epub 2013 Jul 10
[PubMed PMID: 23850902]
[90]
Rothman A, Jonas M, Castel D, Tzafriri AR, Traxler H, Shav D, Leon MB, Ben-Yehuda O, Rubin L. Pulmonary artery denervation using catheter-based ultrasonic energy. EuroIntervention : journal of EuroPCR in collaboration with the Working Group on Interventional Cardiology of the European Society of Cardiology. 2019 Oct 20:15(8):722-730. doi: 10.4244/EIJ-D-18-01082. Epub
[PubMed PMID: 31062694]
[91]
Ciarka A, Doan V, Velez-Roa S, Naeije R, van de Borne P. Prognostic significance of sympathetic nervous system activation in pulmonary arterial hypertension. American journal of respiratory and critical care medicine. 2010 Jun 1:181(11):1269-75. doi: 10.1164/rccm.200912-1856OC. Epub 2010 Mar 1
[PubMed PMID: 20194810]
[92]
Velez-Roa S, Ciarka A, Najem B, Vachiery JL, Naeije R, van de Borne P. Increased sympathetic nerve activity in pulmonary artery hypertension. Circulation. 2004 Sep 7:110(10):1308-12
[PubMed PMID: 15337703]
[93]
Juratsch CE, Jengo JA, Castagna J, Laks MM. Experimental pulmonary hypertension produced by surgical and chemical denervation of the pulmonary vasculature. Chest. 1980 Apr:77(4):525-30
[PubMed PMID: 7357977]
[94]
Christie JD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Dobbels F, Kirk R, Rahmel AO, Stehlik J, Hertz MI, International Society of Heart and Lung Transplantation. The Registry of the International Society for Heart and Lung Transplantation: 29th adult lung and heart-lung transplant report-2012. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2012 Oct:31(10):1073-86. doi: 10.1016/j.healun.2012.08.004. Epub
[PubMed PMID: 22975097]
[95]
Yusen RD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Goldfarb SB, Levvey BJ, Lund LH, Meiser B, Rossano JW, Stehlik J. The Registry of the International Society for Heart and Lung Transplantation: Thirty-second Official Adult Lung and Heart-Lung Transplantation Report--2015; Focus Theme: Early Graft Failure. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2015 Oct:34(10):1264-77. doi: 10.1016/j.healun.2015.08.014. Epub 2015 Sep 3
[PubMed PMID: 26454740]
[96]
Olsson KM, Hoeper MM, Pausch C, Grünig E, Huscher D, Pittrow D, Rosenkranz S, Gall H. Pulmonary vascular resistance predicts mortality in patients with pulmonary hypertension associated with interstitial lung disease: results from the COMPERA registry. The European respiratory journal. 2021 Aug:58(2):. pii: 2101483. doi: 10.1183/13993003.01483-2021. Epub 2021 Aug 26
[PubMed PMID: 34385266]
[97]
Zeder K, Avian A, Bachmaier G, Douschan P, Foris V, Sassmann T, Troester N, Brcic L, Fuchsjaeger M, Marsh LM, Maron BA, Olschewski H, Kovacs G. Elevated pulmonary vascular resistance predicts mortality in COPD patients. The European respiratory journal. 2021 Aug:58(2):. pii: 2100944. doi: 10.1183/13993003.00944-2021. Epub 2021 Aug 26
[PubMed PMID: 33986032]
[98]
Chang KY, Duval S, Badesch DB, Bull TM, Chakinala MM, De Marco T, Frantz RP, Hemnes A, Mathai SC, Rosenzweig EB, Ryan JJ, Thenappan T, PHAR Investigators *. Mortality in Pulmonary Arterial Hypertension in the Modern Era: Early Insights From the Pulmonary Hypertension Association Registry. Journal of the American Heart Association. 2022 May 3:11(9):e024969. doi: 10.1161/JAHA.121.024969. Epub 2022 Apr 27
[PubMed PMID: 35475351]
[99]
Vizza CD, Hoeper MM, Huscher D, Pittrow D, Benjamin N, Olsson KM, Ghofrani HA, Held M, Klose H, Lange T, Rosenkranz S, Dumitrescu D, Badagliacca R, Claussen M, Halank M, Vonk-Noordegraaf A, Skowasch D, Ewert R, Gibbs JSR, Delcroix M, Skride A, Coghlan G, Ulrich S, Opitz C, Kaemmerer H, Distler O, Grünig E. Pulmonary Hypertension in Patients With COPD: Results From the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Chest. 2021 Aug:160(2):678-689. doi: 10.1016/j.chest.2021.02.012. Epub 2021 Feb 11
[PubMed PMID: 33581097]
Level 2 (mid-level) evidence
[100]
Saunders H, Helgeson SA, Abdelrahim A, Rottman-Pietrzak K, Reams V, Zeiger TK, Moss JE, Burger CD. Comparing Diagnosis and Treatment of Pulmonary Hypertension Patients at a Pulmonary Hypertension Center versus Community Centers. Diseases (Basel, Switzerland). 2022 Jan 7:10(1):. doi: 10.3390/diseases10010005. Epub 2022 Jan 7
[PubMed PMID: 35076491]