Introduction
Scleroderma is a connective tissue disorder characterized primarily by the thickening and hardening of the skin. The combining form “sclero” means "hard" in Greek, and the word “dermis” means skin. There are two primary types of scleroderma: localized and systemic (also called systemic sclerosis). In localized scleroderma, the disease affects mainly the skin and may have an impact on the muscles and bones. In systemic scleroderma, there is an involvement of the internal organs, such as the digestive tract, heart, lungs, and kidneys, among others. The severity and outcome of scleroderma are variable.[1][2]
Etiology
The causes of scleroderma are not fully known. There is some evidence that genetic and environmental factors play a role in the genesis of scleroderma. Silica and certain organic solvents are recognized as risk factors of occurrence of systemic scleroderma. The result is an activation of the immune system, causing blood vessel damage and injury to tissues that result in scar tissue formation and the accumulation of excess collagen.
Genetic factors play at least a limited role. According to three US cohorts, the prevalence of the disease was 13 times higher in first-degree relatives of scleroderma patients than in the general population. OX40L gene polymorphism correlates with systemic scleroderma. IRF5 gene was found to correlate with systemic scleroderma as well as with the occurrence of interstitial lung disease during scleroderma.[3][4][5][6]
Epidemiology
Scleroderma is a rare disease. Its prevalence varies with ethnicity, gender, and geographic area. In France, the estimated incidence of systemic sclerosis was 158.3 per million in 2002, mostly limited to the cutaneous forms. The prevalence of the disease in Detroit was 242 cases per million adults with an annual incidence of 19.3 new cases per year per million adults over the period 1989-91. As in numerous other autoimmune diseases, women are at higher risk than men (ratio 4.6:1). Systemic scleroderma can occur at any age; however, it is rare in children and the elderly. The disease is most prevalent in individuals aged 30-50 years.
Localized scleroderma affects mostly women with an incidence of 3 cases per 100,000 individuals/year. Plaque form (also called morphea) is more prevalent in adults while linear scleroderma affects mostly children.[1][7][8]
Pathophysiology
Three primary mechanisms contribute to the development of the scleroderma: vascular anomalies, excess fibrosis, and autoimmune phenomenon.
Abnormal interactions between endothelial cells, fibroblasts, and lymphocytes (B and T) lead to microcirculatory vascular involvement. The endothelial cells produce large amounts of endothelin 1, causing vasoconstriction and fibroblast activation. Also, fibroblasts and activated endothelial cells produce reactive oxygen species that speed up vascular remodeling, leading to the obliteration of small vessels. Activated fibroblasts differentiate easily into myofibroblasts, which have increased collagen synthesis ability.[6][9]
History and Physical
There are two primary forms of scleroderma: localized scleroderma and systemic scleroderma also called systemic sclerosis (SSc).
Localized scleroderma
Localized scleroderma is limited to the skin and underlying tissue. It generally evolves in three consecutive phases: edematous, indurated and sclerotic, and then atrophic. Its outcome is unpredictable and spontaneous improvements are possible. Different clinical forms of localized scleroderma can coexist in the same patient. There are two main groups of localized scleroderma: morphea and linear scleroderma.[1][10]
- Morphea is the most common clinical form. It presents as a single or multiple plaques. These plaques are initially erythematous and then become sclerous, white, indurated, surrounded by a characteristic erythematous halo called “purple ring” reflecting its inflammatory activity. It is generally painless, but pruritus may be present. Later, hypopigmentation appears, and the lesions become generally atrophic. The lesions are mainly on the trunk and proximal extremities; the face is rarely affected.
Plaque morphea can further classify into other sub-types, according to the shape or depth of the lesions. “Guttate” morphea or “white spot disease” refers to “drop-like” shaped areas of skin involvement, whereas “subcutaneous” or deep morphea indicates a substantial involvement of deeper tissues with relative sparing of the overlying skin. The subcutaneous type may rarely extend deep into muscle tissues, but this does not indicate internal organ involvement.
Generalized morphea is defined as multiple morphea plaques, confluent or not, affecting more than two anatomical sites.
Bullous morphea is a rare sub-type characterized by blisters or erosions occurring on plaques of morphea.
- Linear scleroderma manifests as thickened and indurated skin bands, which are most often on the face or extremities. It is the most common form of scleroderma in children.
On the scalp or face, linear scleroderma gives an aspect called "en coup de sabre" (a French expression meaning “cut from a sword”). The sclerotic band is generally located on the forehead but can extend to the scalp (causing scarring alopecia), and to the nose as well as the upper lip. The skin is hypo or hyperpigmented, atrophic and adheres to the underlying bone. This form of scleroderma can sometimes be associated with ipsilateral hemiatrophy of the face and is therefore hardly individualizable from Parry-Romberg syndrome.
Linear scleroderma of the limbs is called "monomelic" and often begins in childhood. Sclero-atrophic bands appear gradually and then extend to the muscles and tendons; this may lead to an extreme aspect of pansclerotic morphea with joint and bone deformities sometimes associated with a stop or growth retardation of the limb.
Systemic scleroderma or systemic sclerosis
This form of scleroderma is the most severe since it usually affects several internal organs. However, most patients do not have all the symptoms described below.
Raynaud’s phenomenon
Raynaud’s phenomenon (RP) is secondary to distal arteries vasospasm after exposure to cold or temperature changes. It is the most common manifestation of systemic sclerosis, occurring in more than 95% of patients. The typical RP crisis includes three phases: syncopal pallor with anesthesia limited to a few fingers, cyanosis paresthesia, and late erythematous, painful phase. Trophic disorders, mainly digital ulcerations, often complicate RP. About 50% of systemic scleroderma patients will have during the course of the of their disease at least one episode of digital ulceration. Digital ischemia can eventually result in distal gangrene that may result in auto-amputation. In more than 95% of patients with SSc, nailfold capillaroscopy shows vascular anomalies, such as architectural disorganization, giant capillaries, hemorrhages, loss of capillaries, angiogenesis and avascular areas.[11][12]
Skin manifestations:
Skin lesions are bilateral and symmetrical, with a distal beginning, involving the fingers and sometimes the toes. Two clinical features should be distinguished [12][13]
- Diffuse SSc, during which cutaneous sclerosis rises above the elbows and knees, which represents 30 to 40% of all patients.
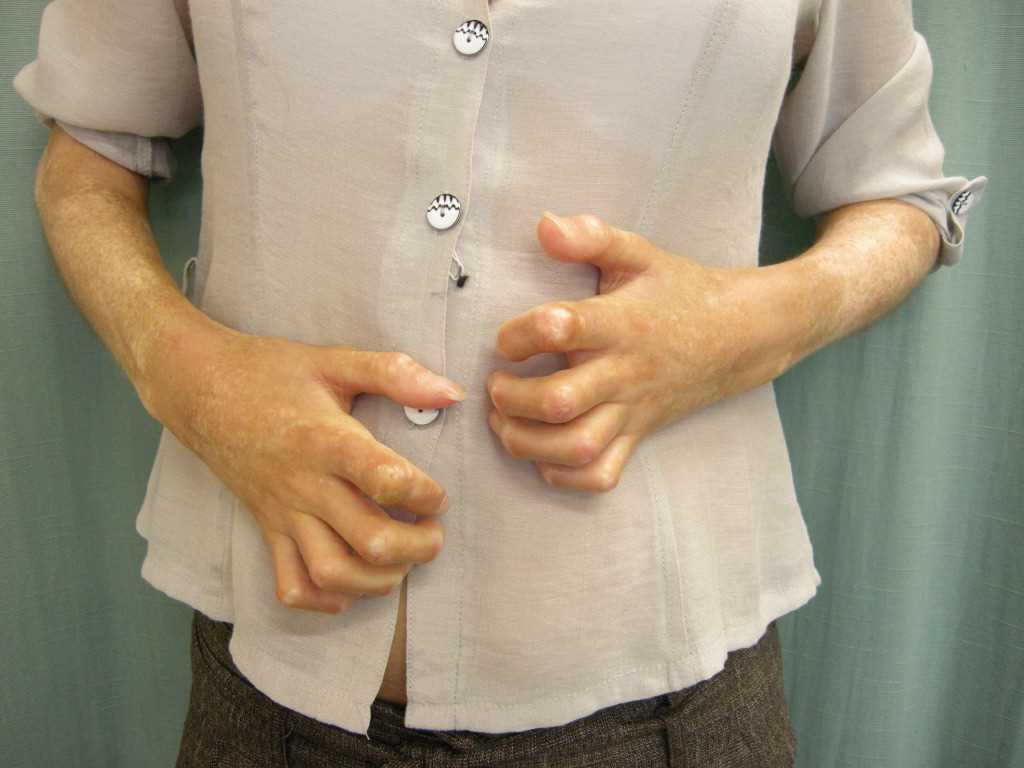
- Limited SSc, during which the cutaneous involvement limits itself to the fingers, hands, forearms, and face. Swelling of the fingers and hands may precede several months the onset of sclerotic lesions. Moreover, the skin of the fingers appears tense, giving a "sausage-like" appearance. The skin of the fingers becomes dry, thick and rough to the touch: this is called sclerodactyly, making it difficult to close the hand into a fist.
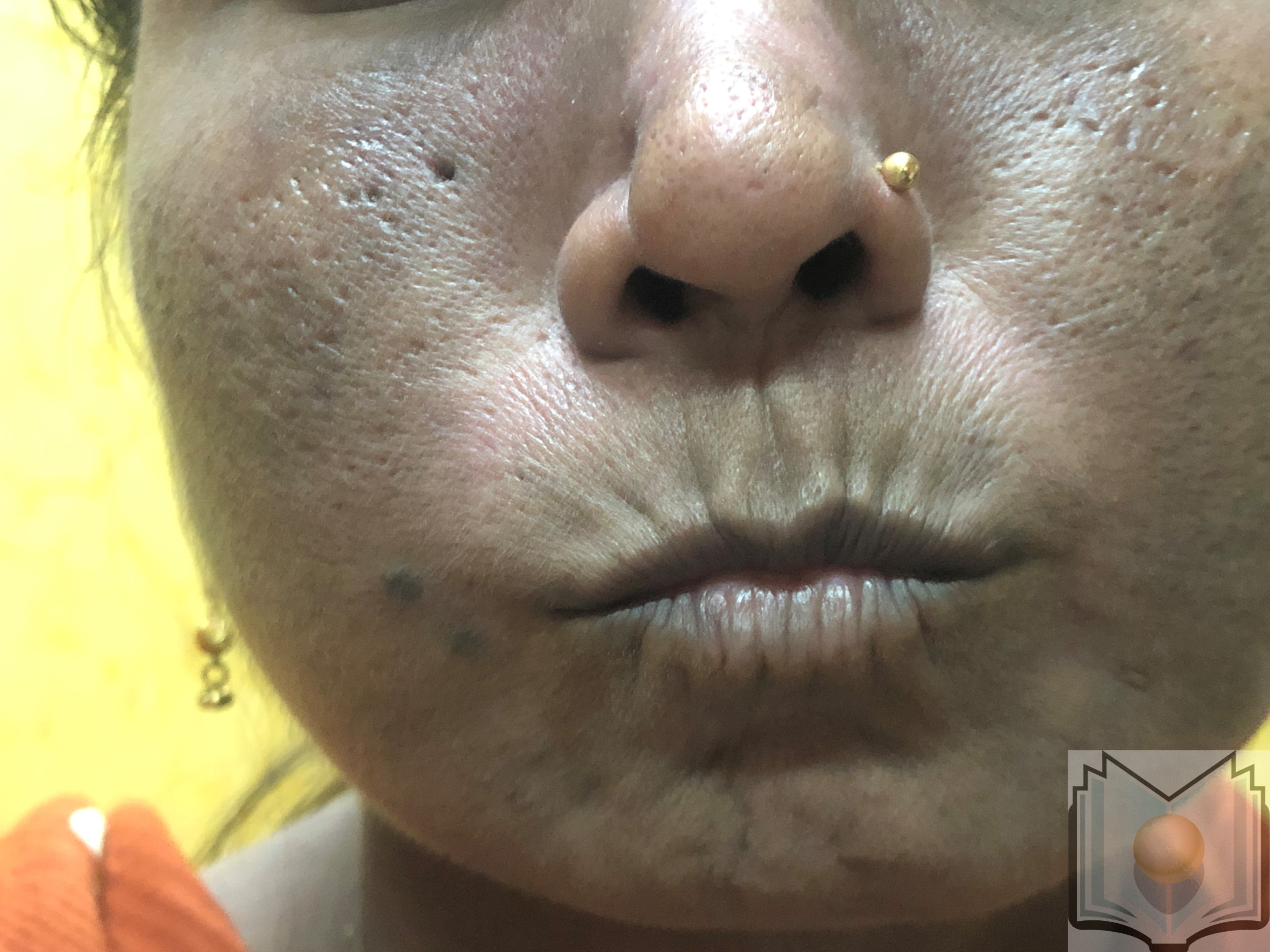
The nails diminish in size, curl and sometimes disappear. The face takes an inexpressive appearance, with a waxy skin in which the folds fade. The nose and the lips thin. Due to skin xerosis, small wrinkles form on the skin around the lips, which become finer. The limitation of oral opening may constitute a limitation for the consumption of solid foods, dental care, and other actions. The ability to open the oral cavity is measured by the inter-incisor distance. Telangiectasias are common during SSc and are usually localized in the face and mucous membranes.
Other cutaneous signs such as calcinosis, pigmentation disorders, and telangiectasia may be present.
Modified Rodnan skin score measures by palpation the extent and importance of cutaneous sclerosis in 17 zones of the body (0: normal cutaneous thickness; 1: minimal thickening; 2: moderate thickening; 3: thickening major). In diffuse forms, this score has a prognostic value, and usually reaches its maximum in the first three years after Raynaud's phenomenon and then tends to improve spontaneously.[14]
Pulmonary impairment
The pulmonary lesions include interstitial lung disease (ILD) and pulmonary arterial hypertension (PAH). These two complications are the leading causes of death during SSc.
- Pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) occurs in up to 13% of patients with systemic scleroderma. It occurs more commonly in patients with limited cutaneous systemic sclerosis. By definition, PAH is pulmonary arterial pressure average greater than or equal to 25 mmHg measured at rest by right catheterization. PAH is suspected when pulmonary arterial systolic pressure exceeds 40 mm Hg at rest in echocardiography. Patients can remain asymptomatic for an extended time, especially if they do not have high levels of physical activity. Syncope, hemoptysis, and dysphonia (Ortner's syndrome) are signs of seriousness. An examination may find a systolic murmur of tricuspid insufficiency or diastolic murmur of pulmonary insufficiency, a loud second heart sound and signs of right heart failure.[15]
- Interstitial lung disease (ILD)
Interstitial lung disease (ILD) is common in patients with SSc since up to 90% of patients exhibit evidence of interstitial changes on high resolution computed tomography (HRCT). It is more frequent with the diffuse forms of the disease. Pulmonary auscultation reveals crackles in the bases with or without a dry cough and dyspnea. ILD assessment is by pulmonary function tests (PFTs) and HRCT. The most classic finding is the ground-glass, and then appear linear or reticular images, septal or intralobular, then a honeycomb appearance with traction bronchiectasis. SSc-ILD appears in the early years of the disease, and its most marked progression occurs in the first years after diagnosis. ILD progression is usually slow; however, about 12% of scleroderma patients will progress to terminal respiratory failure. An ILD involving more than 20% of the lung parenchyma correlates with poorer survival.[16][17][18]
Gastrointestinal manifestations
Gastrointestinal involvement in SSc concerns the whole digestive tract. Gastroesophageal reflux disease (GERD) occurs in 75 to 90% of patients. If a patient complains from dysphagia, erosive esophagitis, peptic stenosis, and endo-brachy-esophagus should be ruled out. Motor impairment can also affect the stomach, causing gastroparesis and more rarely watermelon stomach, a condition related to gastric antral vascular ectasia. In the small bowel, the motor impairment leads to bacterial overgrowth, with concomitant nutritional deficiencies (folate and vitamin B12), malabsorption (steatorrhea), and pseudo-obstruction. Anorectal involvement may cause fecal incontinence and rectal prolapse.[19]
Cardiac manifestations
They are common in SSc. However, only 15% of patients are symptomatic and then have a poor prognosis (estimated 2-year mortality: 60%). Cardiac involvement includes myocardial disease, conduction system defects, arrhythmias, or pericardial disease.
Usually, only the coronary microcirculation is affected. In the case of pulmonary disease, right heart failure is possible. Pericardial involvement is quite common during SSc, usually without clinical consequence. Conduction disorders are quite rare. Rhythm disorders are detectable by Holter-electrocardiography.[20][21]
Renal involvement
Renal involvement is common in patients with SSc. The most serious manifestation is scleroderma renal crisis (SRC) which is a rare but major complication. It characteristically demonstrates the occurrence of sudden-onset hypertension and oligo/anuric acute renal failure. Clinical signs are those of malignant hypertension. SRC is more common in patients with diffuse SSc and having anti-RNA-polymerase III antibodies. Before the use of angiotensin-converting enzyme (ACE) inhibitors, SRC was the most common fatal complication of systemic scleroderma.[22][23]
Musculoskeletal Manifestations
Scleroderma may affect joints (arthritis, arthralgia), tendons (rubs, tenosynovitis) and muscles (myalgia, weakness, more rarely myositis). Friction tendon rubs, seen on the hands, knees, and ankles, indicate a poor prognosis. Digital retractions in flexion due to skin sclerosis and calcinosis, lead to major functional disorders.[24]
Evaluation
In patients with systemic scleroderma, the clinical assessment has two objectives: to confirm the diagnosis and to look for complications.
Complementary exams to be performed if systemic scleroderma is suspected are:
- Nailfold capillaroscopy
- Screening for antinuclear antibodies (mainly anti-centromere and anti-scl70/anti-topoisomerase antibodies)
- Transthoracic echocardiography
- HRCT of the chest
- Diffusing capacity of the lung for carbon monoxide (DLCO) and spirometry
- Hand X-ray
- Esophageal manometry
When the diagnosis of systemic scleroderma is established, in addition to previous exams, it is necessary to perform:
- Electrocardiogram
- Complete blood count
- Renal and liver functions
- Urinalysis
- NT-pro-BNP dosage
- Upper gastrointestinal endoscopy
Exams that should be done for follow-up each year are:
- NT-pro-BNP dosage
- Spirometry and DLCO
- Transthoracic echocardiography
Treatment / Management
Systemic Scleroderma
Treatment aims to relieve symptoms and to slow disease progression. No treatment can cure the disease. Therapeutic strategies should have their basis on a person’s symptoms and the need to prevent complications. The use of different immunosuppressive drugs remains disappointing. Immunosuppressive therapy is reserved for diffuse SSc of recent diagnosis (less than 3 to 5 years) and patients with progressive PID. Its basis is on induction with cyclophosphamide then a relay by mycophenolate mofetil.
The treatment of Raynaud's phenomenon is with calcium channel blockers. Protection against cold and smoking cessation are essential preventive measures. Patients should avoid drugs that worsen RP, in particular, beta-blockers (even as eye drops). In severe forms with trophic disorders, intravenous iloprost is a possible option. Bosentan has demonstrated efficacy in secondary prevention of digital ulcers.
The treatment of esophageal involvement is with prokinetic agents and proton pump inhibitors. Treatment of diarrhea related to microbial overgrowth is with an antibiotic such as amoxicillin, norfloxacin or sulfonamides.
ACE inhibitors have a significant place in the treatment of renal crisis but they are not recommended as prevention.
As for other forms of PAH, scleroderma-associated PAH may be treated with drugs designed to relax the walls of the pulmonary arteries, or vasodilation. Oxygen therapy and anticoagulants may be useful. Diuretics can help reduce swelling.
Corticosteroids may be useful in certain inflammatory manifestations of the disease as inflammatory joint manifestations and inflammatory myopathies.[11][15][16][16] High doses of corticosteroids may induce scleroderma renal crisis.[17][19][22]
Localized scleroderma
Topical corticosteroids are the main treatment for plaque morphea. Generalized morphea may be treated with combination therapy of systemic steroids and methotrexate or with phototherapy. Linear scleroderma of the face or limbs generally requires the combination of systemic corticosteroids and methotrexate to avoid functional and/or esthetic disabilities.[1]
Prognosis
Life expectancy in patients with systemic sclerosis depends on the extent and severity of internal organ involvement. During SSc, the rate of death is 3.5 times higher compared to healthy subjects of the same age. The overall survival of patients with SSc is in the range of 75 to 80% after five years, 55% after ten years, 35 to 40% after 15 years and 25 to 30% after 20 years. Factors correlating with a higher risk of death are advanced age at diagnosis, male patients, diffuse skin involvement, and visceral involvement (pulmonary, cardiac, renal). The pulmonary involvement is the main prognostic factor: five-year survival is greater than 90% in the absence of ILD and about 70% if ILD is present. The one-year survival is 55% in PAH associated with SSc. Autoantibodies also have a prognostic value: survival after ten years is 93% in the presence of anti-centromere antibodies, 66% in patients with anti-Scl70 and only 30% in patients with anti-RNA-polymerase III. The use of angiotensin-converting enzyme (ACE) inhibitors has transformed the prognosis of SRC, which remains an important cause of death.[7][26][27][28][29]
In localized scleroderma, the hardening of the skin generally ceases in the two years after the onset of the disease, and the lesions do not extend to other parts from the body. However, the disease can sometimes last several years, and some plaques may become more marked even after the end of inflammation.
Enhancing Healthcare Team Outcomes
An interprofessional approach should include several medical specialties (dermatology, internal medicine, rheumatology, cardiology, pulmonology, gastroenterology, nephrology, radiology, biology) for the diagnosis and the treatment.
Because of the musculoskeletal impairment frequently observed in scleroderma patients, kinesitherapy is often needed to prevent definitive damages.