
Introduction
Sweet syndrome, first described in 1964 by Robert Douglas Sweet, is an acute febrile neutrophilic dermatosis.[1] Neutrophilic dermatoses consist of a group of noninfectious disorders that are characterized by neutrophilic infiltration of the skin (epidermis, dermis, or hypodermis) with or without true vasculitis. Neutrophilic dermatoses can be idiopathic or secondary to an underlying disorder, localized or generalized, and may or may not have extracutaneous manifestations. Sweet syndrome belongs to the nonvasculitic group of neutrophilic dermatosis disorders, with others including pyoderma gangrenosum, pustular psoriasis, reactive arthritis (Keratoderma blennorrhagicum), Bowed-associated dermatosis-arthritis syndrome, rheumatoid neutrophilic dermatosis, Behcet disease, acne fulminans, familial Mediterranean fever, and SAPHO syndrome (synovitis, acne, pustulosis, hyperostosis, and osteomyelitis).
Sweet syndrome characteristically demonstrates the sudden onset of well-defined tender plaques or nodules accompanied by fever, arthralgias, ocular inflammation, headaches, and, rarely, oral or genital lesions.[2][3][4] This condition may also be associated with other extracutaneous systemic manifestations, which are, however, rare. The goal of pharmacotherapy in acute febrile neutrophilic dermatosis (eg, Sweet syndrome) is to reduce morbidity and complications. The best first-line option is systemic or topical corticosteroids if the lesions are limited. If corticosteroids are contraindicated, anti-inflammatory medications such as colchicine or dapsone are available options.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Sweet syndrome may be idiopathic, which is most common, or may be associated with an underlying condition.[5][6] Several malignancies have reported correlations with Sweet syndrome, including myeloproliferative disorders (myelodysplasia, acute myelogenous leukemia, chronic myelogenous leukemia, multiple myeloma, monoclonal gammopathy, lymphoma, and rarely with solid tumors. Sweet syndrome may occur before, at the time of, or after the diagnosis of malignancy. When associated with malignancy, this condition is more likely to be associated with older age of onset and cytopenias. Pathological features with the histiocytoid sweet syndrome are more likely to be associated with malignancy, especially myelodysplastic syndrome.
Sweet syndrome can also occur in association with several inflammatory and autoimmune diseases. These include inflammatory bowel disease (ulcerative colitis or Crohn disease), rheumatoid arthritis, systemic lupus erythematosus, Sjogren syndrome, Hashimoto thyroiditis, Behcet disease, and dermatomyositis.[7] Post-infectious Sweet syndrome has been described 1 to 3 weeks after upper respiratory and gastrointestinal infections. Association with other infections such as human immunodeficiency virus, viral hepatitis, tuberculosis, and there have been some reports of chlamydia infection.
Drug-induced Sweet syndrome has been a well-known entity, with several drugs reported to cause Sweet syndrome. Granulocyte colony-stimulating factor is the most common drug, while several others, including antibiotics (minocycline, nitrofurantoin, trimethoprim-sulfamethoxazole, norfloxacin, ofloxacin), antihypertensives (hydralazine, furosemide), nonsteroidal anti-inflammatory drugs (diclofenac, celecoxib), immunosuppressives (azathioprine), antiepileptics (carbamazepine, diazepam), anti-cancer (bortezomib, imatinib mesylate, ipilimumab, lenalidomide, topotecan, vemurafenib), antipsychotics (clozapine), and antithyroid (propylthiouracil). Sweet syndrome, in these cases, occurs after exposure as well as re-exposure to the responsible drug and resolves with or without corticosteroids after the withdrawal of the drug. Pregnancy carries correlations with the development of Sweet syndrome in about 2% of cases. The prognosis is favorable, with spontaneous resolution after delivery and no infant or maternal morbidity or mortality.[8]
Epidemiology
Sweet syndrome has a predilection in women with a woman-to-man ratio of 4 to 1. The typical age of onset is between 30 and 60, although cases have been reported in the pediatric and elderly populations as well. There has been no observed racial predilection.
Pathophysiology
The exact pathogenesis of Sweet syndrome is not known. Some genetic factors such as HLA-B54 in the Japanese population, MEFV gene mutation in familial Mediterranean fever patients, and chromosome 3q abnormalities have been observed in patients with Sweet syndrome.[9][10][11][12] Theories regarding the pathogenesis of Sweet syndrome include hypersensitivity to eliciting bacterial, viral, or tumor antigens that may trigger neutrophil activation and infiltration, leading to Sweet syndrome. Another theory suggests the role of cytokines and chemokines such as granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, interleukin-1, and interferon-gamma with a higher level of these cytokines reported in patients with Sweet syndrome.[13][14] Studies for circulating immune complexes, complement activation, and tissue-bound immunoglobulins have been negative and not shown to be pathognomic in Sweet syndrome.
Histopathology
Histologic findings are a hallmark of Sweet syndrome, and typical histopathological features are a requirement for the diagnosis of Sweet syndrome. The classic histopathologic pattern of acute febrile neutrophilic dermatosis (Sweet syndrome) consists of a diffuse dense neutrophilic infiltrate in the reticular dermis. Leukocytoclastic nuclear debris presents interstitially, and papillary dermal edema is common. True vasculitic changes usually are absent, although subtle vasculitic changes may occur. Eosinophils and lymphocytes may be present, but neutrophils predominate. The epidermis typically is spared, although spongiosis and subcorneal pustule formation may be present.
Direct immunofluorescence testing is not contributory. In rare instances, the inflammation may extend to involve the subcutis. In most instances, cases with subcutaneous involvement show extensive involvement of the reticular dermis. Histiocytoid Sweet syndrome has been described as a variant and includes histiocytoid cells, which are immature myeloid cells, commonly mistaken as histiocytes. The most important differential diagnosis for this variant is leukemia cutis.
History and Physical
While the characteristic presentation is that of an abrupt onset of multiple tender, erythematous plaques or nodules, several extracutaneous manifestations may occur.
Various Manifestations
Cutaneous manifestations
The lesions of Sweet syndrome have an abrupt onset of tender erythematous plaques or nodules in varying sizes in an asymmetric distribution. Pustular, vesicular, bullous, and targetoid lesions can present. Lesions are most commonly seen on the upper extremities, but they can also be present on the face, neck, chest, back, and lower extremities. Oral or genital lesions are rare, with rare cases of oral ulcers reported, especially in association with hematological malignancies. Bullous lesions, which are more commonly associated with a hematological malignancy, may ulcerate, mimicking pyoderma gangrenosum. Subcutaneous involvement of the fat in Sweet syndrome leads to tender nodules that can mimic erythema nodosum. A positive pathergy test, as seen in Behcet disease, can also be seen in Sweet syndrome.
Constitutional symptoms
Fever is almost always present, especially in drug-induced Sweet syndrome, although it may be absent in 10% to 20% of cases of Sweet syndrome secondary to other etiologies. Arthralgia, myalgia, fatigue, malaise, and headache are other common constitutional symptoms seen in Sweet syndrome.
Extracutaneous manifestations
Joint involvement is common and may occur in 30% to 60% of cases. Arthralgias and non-erosive inflammatory arthritis have been reported. Ocular inflammation, especially conjunctivitis, is also commonly seen. Other ophthalmologic manifestations may include episcleritis, scleritis, keratitis (including peripheral ulcerative keratitis), uveitis, and choroiditis.[15] Reports exist of cases of Sweet syndrome with other systemic involvement, including neutrophilic infiltration leading to myocarditis, multifocal sterile osteomyelitis, alveolitis, pleural effusions, and aseptic meningitis.
Diagnostic Criteria
There are proposals for other clinical diagnostic criteria.[16] These have undergone revision, and the revised criteria include 2 significant findings, which are the presence of (1) sudden onset eruption of tender, painful plaques or nodules and (2) neutrophilic infiltrate in the dermis without vasculitis. Both must be present to establish a diagnosis of Sweet syndrome. For diagnostic confirmation of Sweet syndrome, at least 2 minor clinical features should be present.
These features may include the following:
- Fever >38 ºC
- Illness preceded by an upper respiratory or gastrointestinal infection, or associated with an underlying typical inflammatory disorder, malignancy, or pregnancy
- Elevated white cell count with neutrophil predominance and elevated inflammatory markers
- Positive response to corticosteroids
Evaluation
Laboratory findings in patients with Sweet syndrome include elevated markers of inflammation, including erythrocyte sedimentation rate and C-reactive protein, peripheral leukocytosis, and neutrophilia. Biopsy findings of dense can support the diagnosis of infiltrates in the superficial dermis composed primarily of polymorphonucleocytes and marked edema of the dermal papillae. Lymphocytes may also be present in the inflammatory infiltrate.
Besides the laboratory evaluation to aid the diagnosis of Sweet syndrome, workup shall include determining the underlying cause if present, especially malignancies. Complete blood counts with findings of cytopenias, constitutional symptoms including weight loss, lymphadenopathy, and absence of other typically-associated conditions, raise the suspicion of malignancy. Age-appropriate cancer screening, including colonoscopy, mammogram, and Papanicolaou smears, should be pursued.
Screening for testicular and prostate cancer in men and pelvic ultrasound in women should also be considerations for diagnostic workups. Computed tomography scans of the chest, abdomen, and pelvis are also options. Besides malignancy workup, a pregnancy test is necessary for women of childbearing age with Sweet syndrome. Sweet syndrome is usually not an initial presenting feature of autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, Sjogren syndrome, or Behcet disease, and the clinician should pursue diagnostic evaluation for these conditions if other typical clinical findings create suspicion for their presence.
Treatment / Management
In the absence of associated malignancy or inflammatory bowel disease, Sweet syndrome is usually highly steroid-responsive and self-limiting. A 2 to 4-week tapering course of oral prednisone starting at a daily dose of 40 mg to 60 mg is usually effective. Intralesional corticosteroid injections and topical corticosteroids can also be used, especially in localized Sweet syndrome. In cases of recurrence of disease after corticosteroid tapering, several steroid-sparing agents have reportedly shown efficacy, including potassium iodide, colchicine, dapsone, isotretinoin, methotrexate, doxycycline, indomethacin, chlorambucil, and cyclosporine.[17][8][18]
Differential Diagnosis
The primary differential diagnosis of Sweet syndrome is infections given the presence of fever, leukocytosis, neutrophilia, elevated inflammatory markers, and neutrophil-dense infiltrates. Bacterial, fungal, and mycobacterial infections all merit consideration, and the clinician must rule these out. The cutaneous lesions of Sweet syndrome may mimic several other conditions, including:
- Allergic contact dermatitis
- Cellulitis
- Behcets disease
- Herpes simplex
- Hypersensitivity drug reaction
- Erythema multiforme
- Erythema nodosum
- Pyoderma gangrenosum
Other diagnoses in the differential include:
- Leukocytoclastic vasculitis
- Leukemia cutis
Prognosis
Most cases resolve, although some persist indefinitely and can cause chronic pain and skin breakdown. Because this condition can be associated with other diseases, including malignancy, the prognosis varies depending on the underlying cause. Recurrence may occur in up to 50% of patients, usually in cases associated with an underlying inflammatory disease or hematologic malignancy.
Complications
Generally, with timely diagnosis and appropriate treatment, the lesions of Sweet syndrome resolve without scarring.
Pearls and Other Issues
Sweet syndrome can present typically and atypically, and the diagnosis can be challenging. A biopsy is always necessary to confirm the diagnosis. Being mindful of the association with hematologic malignancy is essential. Patients with an underlying myelodysplastic syndrome may have the classical cutaneous lesions of Sweet syndrome but with lymphocytic, infiltrate, and atypical mononuclear infiltrate rather than neutrophils (see Image. Underlying Myelodysplastic Syndrome).
In some patients with classical Sweet syndrome, the signs and symptoms of this condition may spontaneously resolve without any form of medical intervention. However, the lesions often persist for many weeks or months in others. Successful management of any malignancy results in clearing the related dermatosis in patients with malignancy-associated Sweet syndrome. Similarly, spontaneous improvement and subsequent resolution of the syndrome typically follow discontinuation of the associated medication in patients with drug-induced Sweet syndrome.
Enhancing Healthcare Team Outcomes
Sweet syndrome is a rare disorder of the skin. However, patients may usually present to the primary care clinician with abrupt onset of tender plaques or nodules, fever, arthralgias, ophthalmologic manifestations, headaches, and, rarely, oral or genital lesions. The role of dermatologists and pathologists is crucial in confirming the diagnosis. The involvement of other subspecialists, including an oncologist, rheumatologist, gastroenterologist, or infectious disease specialist, may be necessary if an underlying associated condition is present.
The goal of pharmacotherapy in acute febrile neutrophilic dermatosis is to reduce morbidity and complications. The best first-line option is systemic or topical corticosteroids if the lesions are limited. If corticosteroids are contraindicated, anti-inflammatory or immunosuppressive medications may be options. The healthcare team can ensure patient compliance, while a pharmacist can help to ensure avoidance of drug interactions. Recommendations include close monitoring and follow-up of the patient.
Media
(Click Image to Enlarge)
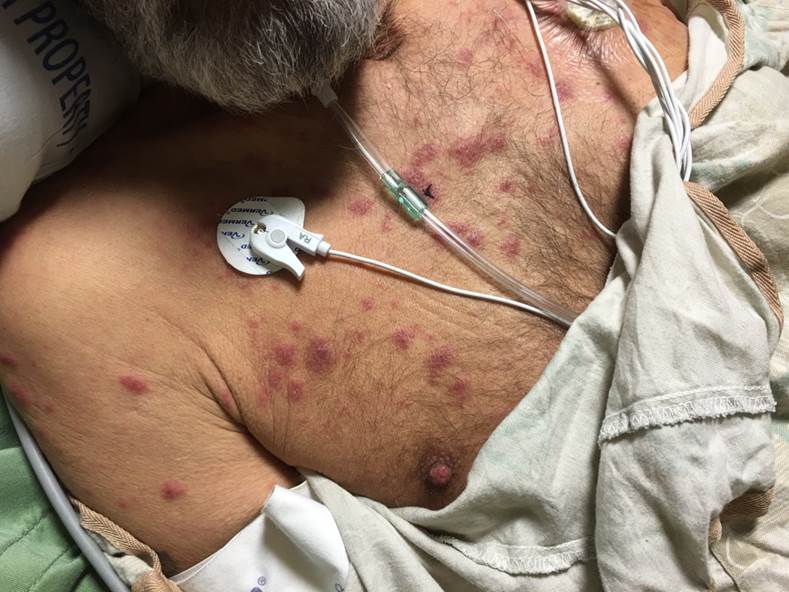
Underlying Myelodysplastic Syndrome. This image shows a patient in the intensive care unit with underlying myelodysplastic syndrome with multiple tender papules and plaques, including lesions with a targetoid and pseudovesicular appearance.
Contributed by S Jones, MD
References
SWEET RD. AN ACUTE FEBRILE NEUTROPHILIC DERMATOSIS. The British journal of dermatology. 1964 Aug-Sep:76():349-56 [PubMed PMID: 14201182]
Bhat AG, Siddappa Malleshappa SK, Pasupula DK, Duke W, Shaaban R. Bullous Variant of Sweet's Syndrome as a Consequence of Radioiodine Contrast Exposure. Cureus. 2018 Oct 24:10(10):e3490. doi: 10.7759/cureus.3490. Epub 2018 Oct 24 [PubMed PMID: 30648032]
Vettakkara KMN, Banerjee S, Mittal A, Goel P, Kumar P, Baitha U, Jorwal P, Soneja M, Biswas A. Not so sweet; severe Sweet's syndrome presenting as SIRS and pleural effusion. Journal of family medicine and primary care. 2018 Nov-Dec:7(6):1584-1587. doi: 10.4103/jfmpc.jfmpc_289_18. Epub [PubMed PMID: 30613566]
Stevens G J, Yutronic H J, Pizarro O J, Velozo P L. Sweet Syndrome in Pediatrics. A case report. Revista chilena de pediatria. 2018 Aug:89(4):511-515. doi: 10.4067/S0370-41062018005000603. Epub [PubMed PMID: 30571826]
Level 3 (low-level) evidenceNelis S, Azerad MA, Drowart A, Lewalle P, Efira A. [Sweet's syndrome induced by pegfilgrastim during a myelodysplastic syndrome AREB2: A case report]. La Revue de medecine interne. 2019 Apr:40(4):258-261. doi: 10.1016/j.revmed.2018.11.007. Epub 2018 Dec 11 [PubMed PMID: 30551891]
Level 3 (low-level) evidenceArun Kumar AU, Elsayed ME, Alghali A, Ali AA, Mohamed H, Hussein W, Hackett C, Leonard N, Stack AG. Sweet syndrome: a rare feature of ANCA-associated vasculitis or unusual consequence of azathioprine-induced treatment. Allergy, asthma, and clinical immunology : official journal of the Canadian Society of Allergy and Clinical Immunology. 2018:14():46. doi: 10.1186/s13223-018-0265-6. Epub 2018 Nov 8 [PubMed PMID: 30455717]
Adil A, Goyal A, Quint JM. Behcet Disease. StatPearls. 2024 Jan:(): [PubMed PMID: 29262080]
Corbeddu M, Pilloni L, Pau M, Pinna AL, Rongioletti F, Atzori L. Treatment of Sweet's syndrome in pregnancy. Dermatologic therapy. 2018 Jul:31(4):e12619. doi: 10.1111/dth.12619. Epub 2018 Jul 25 [PubMed PMID: 30043469]
Takahama H, Kanbe T. Neutrophilic dermatosis of the dorsal hands: a case showing HLA B54, the marker of Sweet's syndrome. International journal of dermatology. 2010 Sep:49(9):1079-80. doi: 10.1111/j.1365-4632.2009.04422.x. Epub [PubMed PMID: 20883277]
Level 3 (low-level) evidenceJo T, Horio K, Migita K. Sweet's syndrome in patients with MDS and MEFV mutations. The New England journal of medicine. 2015 Feb 12:372(7):686-8. doi: 10.1056/NEJMc1412998. Epub [PubMed PMID: 25671271]
Level 3 (low-level) evidenceColović MD, Janković GM, Novak AZ, Strahinja RM, Colović NR. Sweet's syndrome associated with paracentric inversion of chromosome 3q in a patient with multiple myeloma. European journal of haematology. 1996 Aug:57(2):188-9 [PubMed PMID: 8856100]
Level 3 (low-level) evidenceMijovic A, Novak A, Medenica L. Sweet's syndrome associated with inversion of chromosome 3q in a patient with refractory anemia. European journal of haematology. 1992 Sep:49(3):156-7 [PubMed PMID: 1446734]
Level 3 (low-level) evidenceGurnari C, Franceschini L, Anemona L, Passarelli F, Vaccarini S, Pupo L, Provenzano I, Nasso D, Rizzo M, Cantonetti M. Recurrent Sweet's syndrome in a patient with multiple myeloma. Clinical case reports. 2018 Oct:6(10):1958-1960. doi: 10.1002/ccr3.1764. Epub 2018 Aug 21 [PubMed PMID: 30349706]
Level 3 (low-level) evidence. . :(): [PubMed PMID: 30247226]
Gottlieb CC, Mishra A, Belliveau D, Green P, Heathcote JG. Ocular involvement in acute febrile neutrophilic dermatosis (Sweet syndrome): new cases and review of the literature. Survey of ophthalmology. 2008 May-Jun:53(3):219-26. doi: 10.1016/j.survophthal.2008.02.006. Epub [PubMed PMID: 18501268]
Level 3 (low-level) evidenceSu WP, Liu HN. Diagnostic criteria for Sweet's syndrome. Cutis. 1986 Mar:37(3):167-74 [PubMed PMID: 3514153]
Level 3 (low-level) evidenceSanchez IM, Lowenstein S, Johnson KA, Babik J, Haag C, Keller JJ, Ortega-Loayza AG, Cohen J, McCalmont TH, Demer AM, Mansh MD, Hylwa SA, Liu J, Shinkai K. Clinical Features of Neutrophilic Dermatosis Variants Resembling Necrotizing Fasciitis. JAMA dermatology. 2019 Jan 1:155(1):79-84. doi: 10.1001/jamadermatol.2018.3890. Epub [PubMed PMID: 30383110]
Kinser KN, Panach K, Dominguez AR. Recurrent Malignancy-Associated Atypical Neutrophilic Dermatosis With Noninfectious Shock. The American journal of the medical sciences. 2017 Dec:354(6):626-632. doi: 10.1016/j.amjms.2016.10.003. Epub 2016 Oct 20 [PubMed PMID: 29208261]