
Introduction
Gout, once known as the "disease of kings and king of diseases," is among the most prevalent etiologies of chronic inflammatory arthritis in the United States, characterized by monosodium urate (MSU) monohydrate crystals deposition within tissues.[1][2] Hippocrates first described gout in ancient Greece; hence, it is the most understood and manageable disease among all rheumatic diseases.[3][4]
Gout is characterized biochemically by extracellular fluid urate saturation, which is reflected by hyperuricemia in the blood, with plasma or serum urate concentrations exceeding 6.8 mg/dL (approximately 400 µmol/L); this level is the approximate limit of urate solubility.[5] The clinical manifestations of gout may include:
- Acute gout flare (recurrent flares of inflammatory arthritis)
- Chronic gouty arthropathy
- Accumulation of urate crystals in the form of tophaceous deposits
- Uric acid nephrolithiasis
- Chronic nephropathy
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The etiology of gout is usually multifactorial, including genetic predisposition, medical comorbidities, and dietary factors. In rare cases, a single genetic defect may be responsible for causing gout, usually associated with other medical complications. Irrespective of the underlying trigger, the result involves elevated serum uric acid, which can manifest as clinical gout in certain individuals.
Genes Associated with Gout
The heritability of hyperuricemia and gout is about 73% and about 40% to 50% of patients have a family history of gout.[6] Genes associated with gout fall into 4 categories (see Table. Genes Associated With Gout).[6][7]
Table 1. Genes Associated With Gout
| Gene function | Gene name | Gene product | Location |
| Production of uric acid |
HPRT1 PRPS1 ALDH16A1 |
Hypoxanthine guanine phosphoribosyltransferase (HGPRT) Phosphoribosyl pyrophosphate synthetase 1 (PRPPS) Acetaldehyde dehydrogenase 16 family A1 |
Xq26.2-q26.3 Xq22.3 19q13.33 |
| Reabsorption of uric acid in renal tubule |
SLC22A11 SLC22A12 SLC22A13 SLC2A9 |
Organic anion transporter 4 (OAT4) Urate transporter 1 (URAT1) Organic anion Transporter 10 (OAT10) Glucose transporter 9 (GLUT9) |
11q13.1 11q13.1 3p22.2 4p16.1 |
| Excretion of uric acid in renal tubule |
ABCG2 ABCC4 SLC22A6 SLC22A8 SLC17A1 SLC17A3 SLC17A4 SLC2A12 |
ATP-binding cassette transporters G2 (ABCG2) Multidrug resistance protein 4 (MRP4) Organic anion transporter 1 (OAT1) Organic anion transporter 3 (OAT3) Sodium-dependent phosphate transporter 17A1 Sodium-dependent phosphate transporter 17A3 Sodium-dependent phosphate transporter 17A4 Glucose transporter 12 |
4q22.1 13q32.1 11a12.3 11q12.3 6p22.2 6p22.2 6p22.2 6p23.2 |
| Other |
PDZK1 GCKR PKD2 SLC16A9 CARML1 SCGN UMOD ALDH2 |
PDZ domain-containing 1 (scaffolding protein) Glucokinase regulatory protein Ion channels of transient receptor potential superfamily Monocarboxylic acid transporter 9 (MCT9) Myosin 1 connexin (CARMIL) Seceragogin Uromodulin Aldehyde dehydrogenase 2 |
1q21.1 2p23.2 4q22.1 10q21.2 6p22.2 6p22.2 16p12.3 12q24.12 |
Risk Factors
The final step of purine metabolism is the conversion of hypoxanthine to xanthine and then uric acid by xanthine oxidase, which then transforms allantoin by uricase. Allantoin has a much higher solubility than uric acid. Humans, other primates, giraffes, and Dalmatians possess gene mutations that result in the absence of uricase production,[8][9] a genetic mutation resulting in the inactivation of the uricase gene occurred about 25 million years ago. Simultaneously, there was an increase in URAT1 activity, responsible for uric acid excretion. About 20 million years ago, humans and other primates lost the ability to produce vitamin C,[9] leading to the emergence of the antioxidant theory in which uric acid replaced ascorbic acid as the main antioxidant.
One unique aspect of this evolutionary process is the development of hyperuricemia in humans, making them the only known mammals to develop spontaneous gout. Hyperuricemia is the leading cause of gout,[1][10] a condition where uric acid crystals accumulate in joints, causing inflammation and pain. Research has shown that individuals with higher serum urate levels face an increased risk of developing gout and experiencing more frequent flare-ups over time.[11][12] In a study involving over 2000 older adults with gout, those with serum urate levels exceeding 9 mg/dL were 3 times more likely to experience a flare over the next 12 months than those with levels below 6 mg/dL (see Table. Relationship Between Serum Uric Acid Concentration and Incident Gout).[13]
Table 2. Relationship Between Serum Uric Acid Concentration and Incident Gout [12]
| Baseline serum urate | Incidence of gout at 3 years | Incidence of gout at 5 years | Incidence of gout at 10 years | Incidence of gout at 15 years |
| <6.0 | 0.21% | 0.33% | 0.79% | 1.12% |
| 6.0 to 6.9 | 0.37% | 0.66% | 1.98% | 3.70% |
| 7.0 to 7.9 | 0.92% | 1.91% | 6.37% | 9.00% |
| 8.0 to 8.9 | 4.00% | 6.94% | 11.32% | 16.28% |
| 9.0 to 9.9 | 8.31% | 14.02% | 24.18% | 35.21% |
| 10.0 or greater | 10.00% | 26.25% | 40.00% | 48.47% |
Hyperuricemia, while a significant risk factor, does not singularly account for the development of gout (see Table. Risk Factors of Hyperuricemia and Gout); only a minority of individuals with elevated uric acid levels actually develop the condition. To assess the impact of diet on uric acid levels, examining the lower physiological uric acid range in species that do not produce uricase becomes essential. Dietary sources that can contribute to hyperuricemia and gout include the consumption of animal food such as seafood (eg, shrimp and lobster), organs (eg, liver and kidney), and red meat (eg, pork and beef). Additionally, beverages like alcohol, sweetened beverages, sodas, and those containing high-fructose corn syrup may also contribute to the onset of this disease.[1][14][15]
Epidemiological studies have reported a rising burden of gout, primarily attributed to lifestyle changes like increased protein consumption and a sedentary lifestyle. These shifts in habits underscore the intricate relationship between modern living patterns and the prevalence of gout in contemporary society.
Additional factors linked to gout and hyperuricemia include older age, male sex, obesity, a purine-rich diet, alcohol, certain medications, comorbid diseases, and genetic predisposition (see Table. Causes of Hyperuricemia). Medications such as diuretics, low-dose aspirin, ethambutol, pyrazinamide, and cyclosporine have been identified as potential contributors to elevated uric acid levels and gout development.
Table 3. Risk Factors of Hyperuricemia and Gout [1][8][16]
| Modifiable risk factors | Nonmodifiable risk factors |
| Hypertension | Age |
| Obesity | Genetic variants |
| Hyperlipidemia | Gender |
| Diabetes mellitus | Ethnicity |
| Cardiovascular disease | |
| Alcohol | |
| Medications altering urate balance | |
| Chronic kidney disease | |
| Dietary factors |
Table 4. Causes of Hyperuricemia
| Clinical disorders leading to urate and/or purine overproduction | Drug, diet, or toxin-induced urate and/or purine overproduction | Inherited enzyme defects leading to purine overproduction (rare monogenic disorders) |
Causes of hyperuricemia due to decreased uric acid clearance |
| Malignancies | Cytotoxic drugs | Glucose-6-phosphatase deficiency (glycogen storage disease, type I) | Diabetic or starvation ketoacidosis |
| Hemolytic disorders | Ethanol | Hypoxanthine-guanine phosphoribosyltransferase deficiency | Lactic acidosis |
| Myeloproliferative disorders |
Fructose (high fructose corn syrup) |
Phosphoribosylpyrophosphate synthetase overactivity | Chronic renal insufficiency of any form |
| Lymphoproliferative disorders | Ethylamino-1,3,4-thiadiazole | Lead nephropathy (saturnine gout) | |
| Tissue hypoxia | Vitamin B12 deficiency | Hyperparathyroidism | |
| Down syndrome | Pancreatic extract | Sarcoidosis | |
| Psoriasis | Excessive dietary purine ingestion | Chronic beryllium disease | |
| Glycogen storage diseases (types III, V, VII) | 4-amino-5-imidazole carboxamide riboside | Hypothyroidism | |
| Obesity | Preeclampsia | ||
| Insulin Resistance syndrome | Effective volume depletion (eg, fluid losses and heart failure) |
Triggers
Any condition leading to changes in extracellular urate concentration has the potential to trigger a gout flare-up. These conditions include various factors such as stress (mainly due to medical illnesses like cardiovascular illnesses, recent surgical procedure, trauma, dehydration, or starvation), dietary choices (such as the consumption of high-purine foods like organ meats or seafood, as well as alcoholic beverages like beer, wine, and spirits), and drugs (including aspirin, diuretics, or even allopurinol).
Dietary Factors That May Lower Serum Uric Acid
Certain dietary practices have been shown to lower serum uric and reduce the risk of incident gout. Higher consumption of meat and seafood is associated with an increased incidence of gout in men. Conversely, increased intake of dairy products is associated with decreased incident gout in men.[17] Additionally, following the Dietary Approaches to Stop Hypertension (DASH) diet has been proven to decrease serum uric acid levels and mitigate the risk of gout.[18][19] Adequate vitamin C intake is associated with decreased serum uric acid and a reduced risk of gout.[20][21][22][23][24] Furthermore, incorporating cherries into the diet has demonstrated a decrease in serum uric acid[25] and a reduced risk of recurrent gout attacks.[18][26]
Epidemiology
Epidemiological estimates depend on the disease definition. A definitive diagnosis of gout is accepted in the presence of monosodium urate monohydrate crystals in the joint fluid or the identification of tophus. However, given the impracticality of identifying gout through these criteria alone, various case definitions have been devised like self-reports, Rome criteria, the New York criteria, the American College of Rheumatology (ACR) criteria, and the 2015 ACR/European League Against Rheumatism (EULAR) criteria. The 2015 ACR/EULAR criteria have a sensitivity of 92% and specificity of 89%, surpassing the accuracy of all previous definitions and ensuring a more precise and reliable diagnosis of gout in epidemiological studies.
In men, serum urate levels typically range from 5 to 6 mg/dL and are usually attained during puberty, with a slight increase in levels due to age alone.[27] Conversely, women exhibit lower serum urate concentrations averaging 1.0 to 1.5 mg/dL, less than men of corresponding ages,[28][29] a difference likely influenced by renal uric acid clearance under the influence of estrogen. Following menopause, urate concentrations in women rise to levels comparable to those in adult men.[30] The gender-based variation in urate concentration affects the clinical differences between women and men at the onset of gout.[31][32]
The prevalence of gout can vary by age, sex, and country of origin. Generally, the prevalence of gout is 1% to 4%. Older age and male sex are 2 common risk factors recognized globally. In Western nations, the prevalence of gout is significantly higher in men (3%-6%) compared to women (1%-2%), with a notable 2- to 6-fold difference. The prevalence of gout rises with age but plateaus after 70 years (see Table. Prevalence by Age Range).
Data from 2007 to 2008 revealed that around 3.9% of US adults received a gout diagnosis.[33] Estimates regarding gout prevalence in the United States range from less than 3 million to over 8 million individuals. The latest estimates suggest a gout prevalence of over 3% among the adult American population.[34][35][36]
Additionally, data based on the National Health and Nutrition Examination Survey (NHANES) from 2007 to 2016 indicate a higher prevalence of gout in African-American individuals than in White individuals in the USA. Among females, gout prevalence is 3.5% in African Americans and 2.0% in White Americans, with an odds ratio (OR) of 1.81. Among males, the prevalence in African Americans is 7.0% and 5.4% in White Americans, with an OR of 1.26. Hyperuricemia was also more prevalent in African American females and males than their White counterparts, with OR of 2.00 and 1.39, respectively.[37]
Table 5. Prevalence by Age Range
| Location/Age (years) |
20-29 |
30-39 | 40-49 | 50-59 | 60-69 | 70-79 | 80-85 | >85 |
| USA | 0.70 | 0.70 | 3.40 | 3.40 | 8.80 | 8.80 | 8.70 | 8.70 |
| Australia | 0.08 | 0.33 | 1.84 | 1.68 | 3.03 | 4.9 | 6.72 | 7.19 |
| Sweden | 0.06 | 0.27 | 0.80 | 1.54 | 2.83 | 4.89 | 6.61 | 7.38 |
| South Korea | 0.03 | 0.20 | 0.59 | 0.85 | 1.15 | 1.59 | 1.90 | 1.49 |
The incidence rates of gout have displayed an upward trend over the past several decades, with a higher incidence observed in men than women and the incidence rising with age. A study conducted in Olmsted County, MN, from 1989 to 2009 revealed increased gout incidence and comorbidities over the 20 years.[38] Similarly, in the United Kingdom, the prevalence of gout experienced an escalation from 1.52% to 2.49% between 1997 and 2012.[39]
Comorbidities
Gout is associated with health risks, including obesity, hypertension (HTN), chronic kidney disease (CKD), diabetes mellitus (DM), hyperlipidemia (HLD), and metabolic syndrome. A study conducted in Olmsted County, MN, highlighted the increased prevalence of various comorbidities in gout patients compared to the general population. The prevalence of obesity (defined as BMI >35 kg/m2) was 29% in gout patients versus 10% in the general population, HTN was 69% versus 54%, CKD was 28% versus 11%, DM was 25% versus 6%, HLD was 61% versus 21%.[38]
Gaining weight during adulthood has been consistently associated with a heightened risk of developing gout.[40][41][42] Studies from the United Kingdom and Germany have revealed associations between gout and various comorbidities, including DM, congestive heart failure (CHF), HTN, myocardial infarction (MI), and obesity. Additionally, the prevalence of comorbidities increased with higher serum uric acid levels.[43]
Other gout-related comorbidities include HLD, hypothyroidism, anemia, psoriasis, chronic pulmonary disease, osteoarthritis, and depression.[44] Due to increased cell turnover in the epidermis, psoriasis leads to elevated uric acid production. At the same time, patients with CKD experience reduced urate excretion, resulting in hyperuricemia and an increased risk of incident gout.[45]
Gout is associated with a heightened risk of ischemic heart disease (hazard ratio [HR] 1.86), MI (HR 3.246), and cerebrovascular disease (HR 1.552).[46] Moreover, individuals with recent gout flares experience a transient increase in cardiovascular events.[47]
Gout is linked to increased overall mortality, encompassing all-cause mortality and specific causes such as cardiovascular disease, infectious disease, and cancer-related deaths.[48] Particularly, gout is strongly associated with elevated cardiovascular mortality [49] and contributes to mortality related to renal disease, digestive diseases, and dementia.[50]
The connection between gout and dementia, including Parkinson disease, is complex and not fully understood. Studies have shown varied associations, with some indicating a lower risk of dementia,[51][52][53] specifically Alzheimer disease,[54][55] in individuals with hyperuricemia and gout. However, conflicting data suggests that hyperuricemia and gout are associated with an increased risk of dementia.[56][57] Similarly, the relationship between gout and Parkinson disease is inconclusive, with studies showing differing results, including lower,[58][59][60] no specific,[61][62] or a higher risk of Parkinson disease in patients with gout.[63]
Pathophysiology
Gout is an inflammatory arthritis triggered by the deposition of MSU crystals, the end product of human purine metabolism, in joints, soft tissues, and bones. This condition may manifest in many forms, including acute gout flare (acute arthritis), chronic gouty arthritis (chronic arthritis), tophaceous gout (formation of tophi), renal functional impairment, and urolithiasis.[64][65][66]
The pathophysiology of gout involves a series of complex and interacting processes as follows:[67]
- Various genetic and metabolic factors contribute to hyperuricemia in the bloodstream.
- Metabolic, physiologic, and other characteristics are responsible for MSU crystal formation.
- Soluble inflammatory factors, cellular elements, innate immune processes, along with the characteristics of MSU crystals, promote an acute inflammatory response.
- Immune mechanisms come into play to mediate the resolution of acute inflammation induced by MSU crystals.
- Chronic inflammatory processes coupled with the effects of immune cells and crystals on osteoblasts, chondrocytes, and osteoclasts contribute to cartilage attrition, bone erosion, joint injury, and the formation of tophi.
Uric Acid Physiology
Uric acid is the final product of purine metabolism in humans and higher primate species due to a mutation that silences the gene decoding the enzyme uricase.[8][9] Traditionally, it was believed that uric acid played a crucial role as a natural antioxidant in the human body, primarily responsible for eliminating reactive oxygen species. However, recent studies revealed that uric acid is not a significant factor in controlling oxidative stress. Instead, it is thought to be involved in immune surveillance and regulating blood pressure and intravascular volume.
Uric acid is a weak organic acid that predominantly exists in its ionized form, MSU, at pH 7.4. This form is less soluble due to the high sodium concentration. In acidic environments like urine, uric acid exists in its nonionized form, which is even less soluble within the physiological range. Consequently, uric acid crystals and stones can form in the urinary tract, distinguishing them from MSU associated with gout.[8]
Most urate in the body is produced endogenously in the liver with a minor contribution from the small intestines. Renal excretion is pivotal in managing the body's urate pool under steady-state conditions since the glomerulus filters nearly all urate. In a hyperuricemic state, the urate pool expands.
In men, the normal urate range is 800 to 1000 mg; in women, it ranges from 500 to 1000 mg. Urate turnover ranges from 500 to 1000 mg daily. During male puberty, serum urate concentrations increase to reach the adult range, whereas urate levels remain low in females of reproductive age. This disparity is due to estrogen's impact on renal urate transporters, resulting in less renal urate reabsorption and increased clearance in women. However, in menopausal and postmenopausal women, urate levels approach those of adult males and may be influenced by hormone replacement therapies.[8]
The following distinguishes between causes of lower and higher urate levels:
Lowered urate pool Raised urate pool
| Intestinal excretion (ABCG2) | Renal tubular reabsorption |
| Glomerular filtration | Dietary purines, alcohol |
| Urate-lowering drugs | Metabolic disorders, insulin resistance |
| Weight reduction | Purine salvage pathways |
| Renal tubular secretion | ATP turnover |
Hyperuricemia
Hyperuricemia plays a pivotal role in developing gout as it facilitates the nucleation and growth of MSU crystals by reducing urate solubility. Several factors promote hyperuricemia in humans, like the genetic absence of uricase, the reabsorption of 90% of filtered uric acid, and the limited solubility of MSU and urate in body fluids. An imbalance in the production and excretion of uric acid leads to rising serum uric acid levels.[10] When renal urate excretion is decreased, intestinal uricolysis increases to half of the total urate disposal, with the transporter ABCG2 playing a pivotal role. Serum urate concentrations exceeding 6.8 mg/dL become saturated and increase the risk of crystal deposition. Hyperuricemia affects 20% of adult white men in the US and is associated with several chronic disorders.
Hyperuricemia can occur as either primary (idiopathic) or secondary. Overproduction of uric acid is observed in several diseases, toxic states, and due to certain medications. Examples include acute leukemia, tumor lysis syndrome, and psoriasis.
Purine Metabolism
Purines consist of 9-carbon purine nuclei that form fused pyrimidine and imidazole rings. Purines perform essential functions in all living cells through purine-based nucleic acids, including adenine, guanine, and hypoxanthine. The contribution of dietary purines to the urate pool is significant. Removing purines from the diet of normal individuals for 10 days reduces urate levels by 25% and urinary uric acid excretion by 50%. However, implementing severely purine-restricted diets is impractical. Conversely, diets high in fructose, meat, alcohol, and fish promote hyperuricemia.[17]
The endogenous pathway of purine production, known as de novo purine synthesis, involves the conversion of ribose-5-phosphate from 5-phosphoribosyl 1-pyrophosphate (PRPP) into nucleotide inosine monophosphate through 10 key steps. This energy-intensive process prompts energy conservation through the interconversion and salvage of purine nucleotides. Urate precursors of purine degradation are hypoxanthine and guanine, most of which are salvaged. Unused guanine is deaminated to become xanthine, while hypoxanthine is oxidized to xanthine by xanthine oxidase.[8]
Xanthine oxidase is a flavoprotein containing molybdenum-pterin and iron sulfide clusters. It operates in 2 forms: as an oxidase, utilizing oxygen to convert hypoxanthine to xanthine and then to urate, and as a dehydrogenase, using nicotinamide adenine dinucleotide (NAD+). Inhibiting xanthine oxidase is the primary target for lowering urate levels in patients with gout.
The primary regulatory steps in purine synthesis include:
- The synthesis of PRPP in the PRPP synthetase pathway.
- The utilization of PRPP in the first step of de novo purine synthesis.
The pathway is regulated through inhibition by purine nucleotide products of purine synthesis and activation by increased PRPP. This antagonistic control mechanism is disrupted in 2 rare X-linked disorders: deficiency of the salvage enzyme hypoxanthine-guanine phosphoribosyl transferase (HGPRT) and overactivity of PRPP synthetase (PRS1). Conditions such as excessive adenosine triphosphate (ATP) depletion during tissue hypoxia or acute alcohol intoxication can lead to decreased concentrations of inhibitory nucleotides and excess urate production.
Renal Uric Acid Secretion
In adults, only 5% to 10% of uric acid is cleared compared to creatinine clearance, despite 100% uric acid filtration at the glomerulus. This is because 90% of the filtered uric acid is reabsorbed in the renal tubules. Consequently, individuals with hyperuricemia resulting from impaired renal excretion may exhibit normal urinary urate levels due to impaired uric acid clearance. Through genomic and molecular studies, researchers have identified several renal uric acid clearance transporters. Among these, glucose transporter 9 (GLUT9) and urate anion transoporter 1 (URAT1) strongly affect serum urate levels.[6][66]
Glucose Transporter 9 (GLUT9)
GLUT9, a product of the SLC2A9 genel, functions as a voltage-driven urate transporter responsible for mediating uric acid reabsorption from tubular cells. GLUT9 exists in 2 isoforms: GLUT9L, located on the basolateral side of the proximal renal tubular epithelium, and GLUT9S, located on the apical side. This transporter is also expressed in the hepatocytes and regulates serum urate concentrations through its dual effects in the kidney and the liver. Additionally, GLUT9 facilitates the transfer of glucose and fructose, which could explain the dietary influence of these substances on hyperuricemia. Studies involving mice with a GLUT9 knockout had moderate hyperuricemia, massive hyperuricosuria, and early-onset nephropathy.[6]
URAT1
URAT1, encoded by the SLC22A12 gene, is highly specific for uric acid and influences renal uric acid transport by mediating the exchange of various anions. Mutations in SLC22A12 can lead to hypouricemia, hyperuricosuria, and exercise-induced renal functional impairment. Uricosuric drugs like probenecid, benzbromarone, and lesinurad inhibit URAT1 and increase uric acid excretion. Other urate transporters include ABCG2, NPT1, NPT4, and multidrug resistance protein 4 (MRP4).[6]
Autosomal Dominant Tubulointerstitial Kidney Disease
Tubulointerstitial kidney disease, caused by pathogenic variants in the UMOD gene, is characterized by early-onset hyperuricemia (with or without gout), hypertension, and progressive tubulointerstitial inflammation and fibrosis. This condition leads to end-stage renal disease by the age of 40. Previously known as familial juvenile hyperuricemia nephropathy and medullary cystic kidney disease, most affected patients exhibit a mutation in uromodulin, which encodes the Tamm-Horsfall protein. Uromodulin maintains the integrity of the ascending loop of Henle by forming a gel-like lattice that coats the luminal side of the tubule. Defects in the lattice alter solute fluxes, reducing Na and Cl reabsorption, decreasing extracellular volume, and compensatory enhancement of sodium-dependent urate transport in the proximal tubule.
Extrarenal Urate Excretion
In the intestines, urate excretion is facilitated by the ABCG2 transporter. Studies involving reduced ABCG2 knockout mice revealed that reduced intestinal urate excretion increased serum urate levels. Consequently, hyperuricemia resulting from urate overproduction can be classified as a renal overload type consisting of extrarenal underexcretion and genuine urate overproduction subtypes.
Urate Crystal Formation
The formation of MSU crystals requires sustained supersaturated concentrations of urate. Factors like the presence of particulate seed, local cation concentrations, pH, temperature, and dehydration influence crystal formation (see Table. Factors Influencing Urate Crystal Formation).[66][68][69][70] Immunoglobulin (Ig) G may also facilitate crystal formation and growth in patients with gout. MSU crystals tend to form in the first metatarsophalangeal joint, midfoot, and Achilles tendon. Emerging evidence indicates a connection between osteoarthritis (OA) and sites of MSU crystal deposition. In osteoarthritic joints, cartilage degradation products like chondroitin sulfate lowers urate solubility, promoting nucleation and crystal growth.[68] The solubility of MSU drops rapidly with decreasing temperature, further impacting crystal formation and deposition.[5]
Table 6. Factors Influencing Urate Crystal Formation
| Presence of particulate seed nuclei like cartilage debris, collagen, hyaluronate, chondroitin sulfate |
| Local cation concentrations |
| pH |
| Temperature |
| Dehydration |
| The balance between macromolecular inhibitors and promoters |
| IgM and IgG |
Inflammatory Response
Histopathologic and imaging studies have shown the presence of urate crystals within joints for prolonged periods without causing overt inflammatory reactions. Heavily crystal-laden fluids (urate milk) are sometimes found in uninflamed joints and bursae. The dense urate crystal mass in tophi sometimes reaches massive dimensions with minor inflammation and symptoms until they exert critical compression of surrounding tissues.
The initiation of inflammation in gout involves microcrystals usually shed from preexisting synovial tophi. This is supported by observing acute gout flares with rapid changes in urate concentrations. The initiation of inflammation depends on multiple factors like the crystal size, the proteins and molecules coating them, and the recruitment of inflammatory cells. MSU crystal surfaces can bind to various proteins, including IgG, lipoproteins, and lipids (see Table. Inflammatory Events in Acute Gout Flare).[8]
The IgG conformational changes encourage phagocytosis by cells possessing Fc-y receptors, such as neutrophils and macrophages.[71] IgG also activates the classical complement pathway. MSU crystals can also directly activate the classical and alternative complement pathways,[72][73] leading to opsonization by depositing the complement split product C3b on the crystals. The apolipoprotein coating on the MSU crystals counteracts the opsonic effects of the IgG Fc and complement proteins. Additionally, it inhibits neutrophil stimulation. Thus, the inflammatory potential of the MSU crystals is a balance between the proinflammatory and anti-inflammatory elements coating the crystal surface. In acute gout, neutrophils are the predominant inflammatory cells in the synovial tissue and fluid, contributing significantly to the proinflammatory stimulus.[8]
In patients with asymptomatic tophi, synovial fluid macrophages frequently contain MSU microcrystals, suggesting active engagement with phagocytes without apparent inflammation. Synovial macrophages and blood monocytes mount a vigorous response to MSU crystals compared to well-differentiated macrophages due to the release of TGF-b1. Researchers have studied 2 main mechanisms of MSU crystal interaction with phagocytes.
- Activation of phagocytes leads to lysosomal fusion, respiratory burst, and the release of lysosomal enzymes and inflammatory mediators, including TNF-alpha and IL-8.[67]
- The predominant pathway of cytosolic protein complex activation involves the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome. MSU crystals activate macrophages and monocytes via toll-like receptors (TLR) 2 and 4, resulting in signal transduction by My88, interleukin-1 receptor-associated kinase 1 (IRAK1), and IRAK4. This activation triggers nuclear factor-kB, which activates the NLRP3 inflammasome. The activated NLRP3 inflammasome subsequently recruits caspase-1, which processes pro-interleukin-1Β (IL-1Β) into its active form, IL-1Β. IL-1Β plays an important role in the inflammatory response to gout by promoting vasodilatation, recruiting monocytes, and initiating and amplifying the inflammatory cascade. Additionally, IL-1Β secretion can result in bone and cartilage breakdown. Other cytokines, such as TNF-alpha, IL-6, CXCL8, and cyclooxygenase 2 (COX-2), are also involved in the inflammatory response.[66][67][70][74][75][76]
Unlike most external stimuli that activate inflammatory cells by a carefully coordinated cell surface signal transduction involving a cascade of tyrosine kinase phosphorylation, MSU crystals bypass this process and directly activate second messenger systems.
Table 7. Inflammatory Events in Acute Gout Flare
| 1. Deposition and release of urate microcrystals |
| 2. Proinflammatory coating (IgG, complement) |
| 3. Initial reaction with resident cells |
| 4. Activation of membrane signaling molecules |
| 5. NLRP3 inflammasome activation: IL-1 beta release |
| 6. Release of cytokines and chemokines |
| 7. Activation of endothelial cell adhesion molecules |
|
8. Activation of neutrophils |
| 9. Phagocytosis of crystals by neutrophils |
| 10. Cross-linking of inflammatory signaling proteins |
|
11. Removal of crystal coating and phagolysosomal rupture, enzyme and mediator release |
|
12. Resolution: aggregated NET formation, cytokines TGF-beta, MC-R, PPAR-gamma, and AMPK activation. |
Termination of The Acute Flare
Acute gout is inherently self-limiting, even without intervention, typically resolving spontaneously within a few days to a few weeks. This phenomenon is intriguing, given the similarity in the molecular mediators of inflammation in gout and other arthropathies and the persistence of MSU crystals.
Following MSU crystal ingestion, neutrophils undergo NETosis (neutrophil extracellular traps). These NETs aggregate and densely pack MSU crystals while degrading the proinflammatory cytokines, including IL-Β, TNF-α, and IL-6. The increased vascular permeability after acute synovitis allows increased entry of anti-inflammatory cytokines and crystal-coating molecules like apolipoprotein B (apoB). Coating with apoB and locally produced apoE and transforming growth factor Β (TGF-Β) inhibits neutrophil activation. Systemic anti-inflammatory mediators like melanocortins decrease joint inflammation by the macrophage melanocortin receptors (MCRs), and adenosine monophosphate-activated protein kinase inhibits NLRP3 expression, which inhibits the cleavage of caspase-1 and secretion of IL-1Β.[66][67][74][76]
Advanced Gout
Tophi are deposits of MSU crystals associated with granulomatous inflammation. They are nests of crystals surrounded by a corona zone composed of differentiated macrophages and multinucleated giant cells encased within a fibrous layer. Proinflammatory cytokines like IL-1 and TNF-α are expressed within the corona. Aggregated NETs are also part of the tophus. The tophus is a dynamic chronic inflammatory response to MSU crystal deposition that is complex and organized.
Tophi are primarily found in periarticular, articular, and subcutaneous areas, including cartilage, bone, joints, tendons, and skin, all rich in proteoglycan. The tissue reaction to tophus is generally chronic inflammation and is both adaptive and innate immunity. Few patients with tophaceous gout also present with chronic gouty arthritis (chronic synovitis). There is a close relationship between MSU crystal deposits and the development of cartilage and bone erosions.[77]
Tophi contribute to joint damage and bone erosion in gout.[78] At the bona and a tophus interface, MSU crystal deposits are surrounded by osteoclast-like cells.[79] T-cells within the tophus express the receptor activator of nuclear factor κB ligand (RANKL), continuing to bony erosions. Additionally, urate crystals decrease osteoblasts' function, viability, and differentiation and reduce osteoprotegerin expression. Hence, more osteoclasts and reduced osteoblasts are present at the bone-tophus interphase.
The double-contoured ultrasound sign is observed in the superficial articular cartilage of patients with chronic gout and represents the presence of urate deposits. Urate crystals degrade cartilage matrix by inducing nitric oxide generation and the expression of matrix metalloprotease 3. Consequently, joints with persistent crystals experience ongoing progressive damage in the absence of acute flares.
Histopathology
When examined under polarized light microscopy, MSU crystal deposition is typically described as a rod or long needle-shaped crystal with negative birefringence.[80] When viewed under light microscopy, tophi exhibit distinct zones: the crystalline center, the surrounding corona zone, and the fibrovascular zone. The corona zone contains multinucleated giant cells, histiocytes, and plasma cells.[81]
History and Physical
Gout has been described as a chronic disease characterized by 4 distinct stages.[3]
- Asymptomatic hyperuricemia
- Acute gout attacks
- Intercritical period
- Chronic tophaceous gout.
Asymptomatic Hyperuricemia
The majority of patients with asymptomatic hyperuricemia never develop gout. The risk of an acute gout attack increases with the serum urate level. This stage ends with the occurrence of the first gout attack.
Acute Gout Attack
The initial manifestation of gout is an acute attack of arthritis, usually monoarticular, marked by the abrupt onset of severe pain and swelling. Maximum inflammation occurs within 12 to 24 hours. Gout flares are typically monoarticular, with 85% to 90% of cases occurring in the lower extremities.[3][82] The first metatarsophalangeal joint is the most commonly involved, with about 50% of initial attacks occurring there and 90% of patients experiencing at least 1 attack in this joint.[3] The talar, subtalar, ankle, and knee can also be involved. In 3% to 14% of cases, the initial attack is polyarticular, causing some confusion.[3]
Although affliction of the joints mentioned above is common in gout, healthcare professionals should also consider other joints, specifically those with underlying osteoarthritis. Periarticular structures such as tendons and bursa may also be involved.[1] While gout can occur in axial joints such as sacroiliac joints and the spine, this is much less common than peripheral involvement, leading to diagnostic confusion.[83][84] Acute gouty arthritis may be associated with fever and leukocytosis, making it difficult to differentiate from septic arthritis. The initial attack resolves within 3 to 14 days, even without pharmacotherapy. Over time, gout flares occur more frequently, become less intense, and involve more joints.[3]
Polyarticular gout flares are more likely to occur in patients with longstanding disease. Initial presentation of polyarticular gout is more frequent in patients in whom gout and hyperuricemia arise secondary to lymphoproliferative or myeloproliferative disorders or in organ transplant recipients receiving tacrolimus or cyclosporine.[85][86] Additionally, in South Africa, a polyarticular presentation in women, with only a small number starting with acute podagra. Rarely, patients may develop tophi without a history of acute gouty flares.
Gout flares are more common at night and early morning when cortisol levels are low.[87] The pain is often sudden, waking the patient from sleep, or it may have developed gradually over a few hours before the presentation, reaching its maximum intensity of pain at 24 hours.[87] Signs of inflammation may extend beyond the joint involved, giving the impression of cellulitis with erythema and desquamation or dactylitis (sausage digit). The pain is usually severe and not responsive to usual home remedies; even touching the joint can be excruciatingly painful. Gout flare-ups often incite local inflammation, which presents as erythematous, swollen, and warm joints. The erythema over the affected joint during an attack is characteristic of gouty synovitis. Systemic inflammatory features may include fever, malaise, and fatigue.[1]
Around 60% of the patients experience a second attack within 1 year, and 80% within 3 years. Acute attacks can be precipitated by local trauma, alcohol binges, overeating or fasting, weight changes, use of diuretics, and initiation of urate-lowering drugs. In a hospital setting, postoperative status or acute severe medical illnesses such as myocardial infarction, exacerbation of congestive heart failure, or cerebrovascular accident may precipitate attacks.[3] It is not unusual for patients to receive urate-lowering therapy (ULT) during hospitalization, possibly leading to a gout flare. Additionally, the spring season has reportedly been associated with increased gout attacks.
Physical examination findings in gout align with the patient's history. The affected joint is typically red, swollen, warm, and tender.[88] The flare-up in patients with chronic gout may involve multiple joints, causing a systemic inflammatory response syndrome that may mimic sepsis.[89] Tophi, subcutaneous deposits of urate which form nodules, can be found in patients with persistent hyperuricemia. Tophi typically occur in the joints, ears, finger pads, tendons, and bursae.[1]
Intercritical Gout
After resolving the acute attack, the patient enters the intercritical stage. Patients typically feel well during this stage without experiencing joint pain or swelling. Despite the apparent inactivity of the disease, hyperuricemia persists, and crystal deposition continues. Subclinical inflammation may be present in the joints during this period.
Chronic Tophaceous Gout
Patients with gout who are untreated or undertreated may develop chronic tophaceous gout over several years, leading to gradual progressive joint destruction. Gouty tophi, which are foreign bodies surrounded by granulomas containing deposits of MSU crystal, manifest as chalk-like subcutaneous nodules beneath transparent skin with increased vascularity. These nodules may or may not drain. While some patients may present with tophi as their initial symptom, chronic tophaceous gout usually develops 10 or more years after an acute attack. However, microtophi can be observed early in the disease, especially in patients with hyperuricemia. MSU crystal deposition is evident in joints affected by osteoarthritis, primarily in the connective tissue and articular cartilage.
Tophi may occur intraarticularly, periarticularly, or extra-articularly, with common sites including the digits of hands and feet, knees, and the olecranon bursa. This condition leads to destructive deforming arthritis, extensive bone destruction, and severe deformities. Women develop tophaceous deposits on the Heberden nodes and Bouchard nodes. Finger pad tophi were observed in 30% of patients with chronic tophaceous gout. Postmenopausal women with CKD may exhibit finger pad tophi before the onset of an acute attack.[3][2][66]
Tophaceous deposits have been documented in various uncommon sites, including the eye cornea and heart valves. These deposits highlight the systemic nature of gout and its potential to affect diverse tissues and organs beyond the joints.
Evaluation
Synovial Fluid Analysis
Monosodium urate crystal identification remains the gold standard for diagnosing gout.[80] Gout flares are marked by MSU crystals in synovial fluid obtained from affected joints of bursas, visualized through direct examination of a fluid sample using compensated polarized light microscopy. The crystals are often intracellular, indicating active phagocytosis. This technique can also identify uric acid crystals from tophaceous deposits and joints during the intercritical period.[90] During a gout flare-up, synovial fluid is usually yellow and cloudy, containing crystals and white blood cells (WBCs) with neutrophil predominance.
In patients with septic arthritis, the synovial fluid will be more opaque, with a yellow-green appearance. Microscopic examination reveals a higher WBC count (>50000/microL) with a predominance of neutrophils. However, there is considerable overlap in WBC counts and neutrophil percentages between patients with acute gouty arthritis and septic arthritis, making these parameters unreliable for diagnosis. Positive synovial fluid gram stains and cultures, along with low synovial fluid glucose levels, are common findings in septic arthritis. It is essential to note that the presence of crystals in synovial fluid analysis does not rule out septic arthritis, as both conditions can coexist.[91][92]
Under polarizing microscopy, synovial fluid or tophus aspiration analysis reveals needle-shaped, negatively birefringent crystals.[1][3][93] Arthrocentesis is essential to confirm the diagnosis and differentiate it from other conditions such as septic arthritis, Lyme disease, or pseudogout (caused by calcium pyrophosphate crystals).[93]
Laboratory Study
The examination usually reveals elevations in the WBC, erythrocyte sedimentation rate (ESR), and C-reactive protein (CRP) during acute gouty arthritis. These features are nonspecific and do not confirm or differentiate the diagnosis from septic arthritis.[91]
During acute gouty arthritis, the serum urate level may be high, normal, or low. About 50% of patients with acute gouty arthritis will not have an elevated serum uric acid. Serum uric acid measurement during an acute attack is of no diagnostic value; it is most useful when checked after the resolution of the flare. Hyperuricemia is helpful in the clinical diagnosis of gout in symptomatic patients, but hyperuricemia alone does not confirm the diagnosis. Asymptomatic hyperuricemia is not uncommon in the general population. Persistently low serum uric acid concentrations make the diagnosis of gout unlikely.[3] In patients suspected of gout based on clinical features, an elevated serum uric acid level (>6.8 mg/dL) can support the diagnosis but is neither diagnostic nor required to establish the diagnosis. The most accurate time to assess serum urate level to establish a baseline value is 2 weeks or more after a gout flare has subsided.
Urinary fractional excretion of uric acid can be measured, especially in young populations with nonspecific causes of hyperuricemia. The measurement can help differentiate between overproduction or underexcretion of uric acid and can guide therapy.
Imaging
Although not routinely used, ultrasonography and dual-energy CT (DECT) can assist in diagnosing gout.[94][95][96][97] On ultrasound, MSU deposition appears as a hyperechoic enhancement over the cartilage, known as the double contour sign. DECT can identify urate deposits based on the beam attenuation after exposure to 2 different X-ray spectra.[1][3] In a pooled analysis, the ultrasound double contour sign had a sensitivity of 83% and a specificity of 76%, while DECT had a sensitivity of 87% and a specificity of 84% for diagnosing gout.[95] A meta-analysis of ultrasound's diagnostic accuracy, which included features like the double contour sign, tophus, or bony erosion, showed a sensitivity of 65.1% and specificity of 89% for a diagnosis of gout.[98]
Treatment / Management
Specific goals guide the treatment of gout. During acute flares, the primary objective is to alleviate inflammation and symptoms. In the long term, the goal shifts toward reducing serum urate levels to suppress flare-ups and regression of tophi.[3][99][100]
General Principles of Therapy
- Early on, introducing treatment for a gout flare leads to a more rapid resolution of symptoms.
- The duration of gout flare therapy ranges from a few days to several weeks, depending on the timing of treatment initiation.
- Anti-inflammatory gout flare prophylaxis should generally be continued during the early months (up to 6 months) of ULT.
- For patients receiving ULT at the time of gout flare, the urate-lowering medication should be continued without interruption as there is no benefit to temporary discontinuation.
- The presence of tophi indicates initiating long-term ULT either during or following the resolution of a gout flare to reverse or prevent joint damage and chronic gouty arthritis.
Acute Gout Flare
The management of acute flares of gouty arthritis aims to decrease inflammation and resulting pain. Treatment should commence within the first 24 hours of onset to reduce the severity and duration of the flare-up if possible.[10] Nonpharmacological management, such as rest with topical application of ice packs [101] can be combined with medications that reduce inflammation.[102] First-line treatments for gout flares are nonsteroidal anti-inflammatory drugs (NSAIDs), colchicine, or systemic glucocorticoids.[103] The length of treatment should be at least 7 to 10 days to prevent rebound flare-ups.[104] Early initiation of NSAIDs may lead to the resolution of the attack with a single dose.(A1)
NSAIDs
NSAIDs are most effective when therapy is initiated within 48 hours of the onset of gout symptoms. Indomethacin and naproxen are the more potent NSAIDs for gout, although many other commonly used NSAIDs exist. NSAID names and dosing are as follows:
- Indomethacin 50 mg 3 times daily
- Naproxen 500 mg twice daily
- Naproxen 500 mg twice daily
- Ibuprofen 800 mg 3 times daily
- Diclofenac 50 mg 2 to 3 times daily
- Celecoxib 200 mg twice daily
Typically, NSAID treatment for gout flare lasts for 5 to 7 days. There is no significant preference for one NSAID over another, but high-dose, fast-acting NSAIDs such as naproxen or diclofenac are options. Indomethacin is not preferable due to its toxicity profile.[10] NSAIDs are usually given in full doses for the first 3 days and then tapered according to the clinical improvement. COX2 selective inhibitors like celecoxib can be used to reduce adverse GI effects.
Contraindications for the use of NSAIDs include active duodenal or gastric ulcer, cardiovascular disease (uncontrolled HTN or CHF), NSAID allergy, and CKD with creatinine clearance (CrCl) of less than 60 ml/minute per 1.73 square meters. Aspirin is not recommended for treating gout flares due to the paradoxical effects of salicylic acid on serum urate levels.[105][106] This paradoxical effect results from uricosuria at higher doses and renal uric acid retention at lower doses (less than 2 to 3 g/day).[107][108]
Oral Glucocorticoids
Glucocorticoids are recommended for gout patients with contraindications to NSAIDs and colchicine, and they are also preferred for patients with renal insufficiency. The initial dose for a gout flare is:
- Prednisolone or prednisone 30 to 40 mg once daily or divided into twice-daily doses until resolution begins. Taper the dose over the next 5 to 10 days.
This has been proven to be at least comparable to NSAID efficacy. High starting doses of systemic steroids (>0.5 mg/kg body weight) are required for acute gout, especially in patients with a polyarticular presentation. A depot preparation for triamcinolone (60mg once) or methylprednisolone has been reported to be effective.[109][110] However, the dose may need to be repeated at intervals of 48 hours to achieve resolution of the flare. Glucocorticoids can be administered intra-articularly for a monoarticular gout flare-up or orally for polyarticular flare-ups. The efficacy of glucocorticoids is similar to or superior to other agents and has no greater risk of adverse effects in most patients.[111][112][113](A1)
In patients with an unclear diagnosis of an acute gout flare, arthrocentesis and synovial fluid analysis should be performed, and oral and intra-articular glucocorticoids should be avoided until the results are available. Initiation of other agents, such as NSAIDs or colchicine, should be considered. Frequent adverse effects of moderate- to high-dose, short-term glucocorticoid use include hyperglycemia, fluid retention, increased blood pressure, and mood changes. Repeated and regular courses of glucocorticoids should be avoided to limit adverse effects.
In patients with concomitant or suspected infections, uncontrolled diabetes mellitus, prior glucocorticoid intolerance, and post-operative status, glucocorticoids may heighten the risk of impaired wound healing. Careful consideration of these factors is crucial when determining the appropriate course of treatment for patients with gout flares.
Parenteral Glucocorticoids
Intravenous or intramuscular glucocorticoids are suggested for patients who are not candidates for intraarticular glucocorticoid injection or cannot take oral medications. A typical methylprednisolone dose is 20 mg intravenously twice daily, with a stepwise reduction and rapid transition to oral prednisone when improvement begins. Adrenocorticotropic hormone (ACTH) is also efficacious for treating gout flare, but limited availability and high cost restrict its use.
Colchicine
Colchicine, derived from the Colchicum autumnale plant and with a history spanning over 3500 years,[114] has proven comparable in efficacy to other agents when taken within 24 hours of gout flare onset. In a randomized control trial, colchicine reduced pain by over 50% at 24 hours compared to a placebo. The lipophilic nature of colchicine makes it readily bioavailable for cellular uptake after oral administration. The primary target of colchicine is tubulin, and it is metabolized through hepatic elimination.
Colchicine acts by binding tightly to unpolymerised tubulin and forms a colchicine-tubulin complex that regulates microtubule and cytoskeletal function. This regulation extends to diverse cellular processes, including cell proliferation, gene expression, signal transduction, chemotaxis, and neutrophil secretion of granule contents. Furthermore, colchicine decreases neutrophil adhesion by suppressing E-selectin redistribution in the endothelial membrane.
EULAR consensus guidelines recommend a maximum of 3 doses of 0.5mg of colchicine daily for treating acute gout. The total colchicine dose should not exceed 1.8 mg on day 1, 1.2 mg for the first dose followed by 0.6 mg an hour later [US Food and Drug Administration (FDA) approved dose] or 0.6 mg 3 times on the first day.[115] On subsequent days, colchicine should be taken once or twice daily until the gout flare resolves.[116](A1)
A reduced dose of colchicine may be required for patients with diminished hepatic or renal function or those at risk of potential drug interactions. Colchicine toxicity can occur with ABCB1 inhibitors like cyclosporin and clarithromycin; neuromyopathy may develop weeks after initiating cyclosporin. High-dose colchicine regimens should be avoided due to their high toxicity. Adverse effects of colchicine include gastrointestinal (GI) symptoms like nausea and diarrhea, myotoxicity, and myelosuppression (leukopenia, thrombocytopenia, and aplastic anemia).[117] The most common adverse effects are abdominal cramping and diarrhea.[115][118] Intravenous colchicine is strongly discouraged due to serious adverse effects, including death, and it is no longer approved by the FDA in the US.(A1)
Colchicine dosing adjustments for certain high-risk groups of patients should follow the guidelines outlined in the manufacturer's FDA-approved information. Typically, a maximum of 0.3 mg dose is administered on the day of a gout flare, and the dose is not repeated for at least 3 to 7 days or longer in such patients. The high-risk groups include:
- Individuals who have used colchicine prophylaxis in the last 14 days possess normal hepatic and renal function and have taken a medication that inhibits P-glycoprotein or is a potent inhibitor of CYP3A4 within the previous 14 days.
- Individuals who have used colchicine prophylaxis in the last 14 days, regardless of hepatic and renal status, and have taken a medication that is a moderate CYP3A4 inhibitor within the same timeframe.
- Individuals with advanced hepatic or renal impairment (Child-Pugh C cirrhosis or equivalent CrCl of <30 mL/min), regardless of recent colchicine use.
Colchicine exhibits interesting effects beyond treating and preventing gouty arthritis flares. Research suggests it has a beneficial effect on cardiovascular events.[114][119][120] A population study linked colchicine use in patients with gout to reduced cardiovascular events and all-cause mortality.[121] In a randomized, double-blind trial involving post-MI patients within 30 days (n = 4,745), low-dose colchicine use lowered the risk of cardiovascular events (resuscitated cardiac arrest, MI, stroke, and angina leading to revascularization) compared to placebo: 5.5% with colchicine versus 7.1% with placebo; HR 0.77 (95% CI 0.61-0.96; P=0.02).[122] While colchicine did not affect outcomes like death from cardiovascular causes, resuscitated cardiac arrest, or MI, it notably reduced stroke and angina, leading to coronary revascularization.
Prophylaxis For Acute Gout
The subclinical joint inflammation in gout justifies colchicine prophylaxis, as acute gout flares are ULT's most common adverse effect. For prophylaxis, low-dose colchicine therapy is the first choice.[123][124] It is commenced 1 or 2 weeks before using urate-lowering drugs and continues for up to 6 months after normalizing uric acid levels or until the clinically visible tophi are resolved.[124][123] Low-dose NSAIDs and low-dose corticosteroids can be used but carry more toxicity.[125] The recommended colchicine dosage is 0.6 mg once or twice daily without renal or hepatobiliary compromise. In patients with renal impairment, the colchicine dose may be reduced to 0.3 mg daily or 0.6 mg every other day.(A1)
Interleukin-1 Inhibition
IL-1 antagonists have shown efficacy in refractory cases of gouty arthritis. Anakinra, a soluble IL1 receptor antagonist, is administered at 100 mg/day subcutaneously for 3 days [126][127] or a single dose of IL-1 beta monoclonal antibody, canakinumab.[128][129] The subcutaneous dose of 150 mg canakinumab was more effective than a single-dose intramuscular (IM) dose of triamcinolone acetonide, although the risk-benefit ratio is uncertain.(A1)
Urate lowering therapy (ULT)
Non-pharmacologic treatment
Gout is associated with several comorbidities, including obesity.[38] In a study examining the association between obesity and gout, adults aged 40 to 75 years (n = 11,079) in NHANES 2007 to 2014 were categorized into 4 groups: stable obese, weight gain, weight loss, and those maintaining a normal BMI over time (reference group).[40] Among those with stable obesity, the risk of gout was the highest, with an HR of 1.84 (95% CI 1.08-3.14). Patients who gained weight as adults also exhibited an increased risk of gout with HR of 1.65 (95% CI 1.19-2.29).(B2)
Diet can affect serum uric acid levels. Weight loss and dietary adjustments can reduce serum uric acid by 1 to 2 mg/dL. Foods high in purines, such as organ meats, shellfish, and beer, can elevate uric acid levels. Soft drinks containing high-fructose corn syrup are associated with an increased risk of gout;[14][15] therefore, reducing their intake can help reduce serum uric acid. The DASH diet has been proven to lower serum uric acid compared to a standard Western diet, making it beneficial for gout management.[18] Consuming at least 500 mg daily of vitamin C has also been shown to decrease serum uric acid levels and lower the risk of incident gout.[20][21][22][23][24] Studies have shown that higher doses of vitamin C correspond to reduced risk of gout in men.[23] Cherry consumption has also been linked to lowered serum uric acid levels[25] and a decreased risk of recurrent gout attacks[26].(A1)
Pharmacologic
The 2020 American College of Rheumatology Guideline for managing gout [100] advises against initiating ULT after the first episode of acute gouty arthritis. ULT should not be initiated in patients with asymptomatic hyperuricemia. The guidelines provide specific criteria for initiating ULT, including the following:
- Frequent or disabling gout flares (≥2 yearly) that are difficult to treat
- Gout with chronic kidney disease (stage 3 or higher)
- Tophus diagnosis on physical examination or imaging
- Past urolithiasis
- Chronic tophaceous gout
The decision to initiate ULT should be individualized. For instance, in a younger patient with their first gout attack with elevated serum uric acid levels, the likelihood of future gout attacks and progressive joint damage with tophi is higher, making it prudent to start ULT. Conversely, in an elderly patient with gout, multiple comorbidities, and taking multiple medications, the decision to treat may be more nuanced, and careful consideration should be given in this scenario. It is essential to note that the guidelines are to provide guidance but not dictate therapy.
ULT is started at a low dose to monitor the side effects and treatment response. Dose adjustments are made every 2 to 6 weeks to achieve serum urate levels of less than 6 mg/dL or 5 mg/dL in patients with tophi.[100][130] The 2020 American College of Rheumatology Guideline conditionally recommends starting ULT during acute gout flares, with some evidence supporting its safety with medications like allopurinol [131][132] and febuxostat.[133] However, initiating therapy during an acute attack might pose challenges regarding patient compliance, especially considering that patients experiencing acute flares are often hospitalized for the first time.(A1)
During the initiation of ULT, there is an increased risk of gout flare-ups. As a prophylactic measure, colchicine is recommended for 3 months after achieving the serum urate goal in patients without tophi or 6 months in those with tophi. This strategy helps to minimize the risk of flare-ups during this critical period.[100]
ULT can be categorized into 3 classes based on their mechanisms:
Xanthine oxidase inhibitors (XOI)
XOIs work by inhibiting uric acid synthesis. This class includes allopurinol and febuxostat. Allopurinol is the recommended first-line pharmacologic ULT in gout.[100] Physicians should regularly monitor liver enzymes, renal function, and blood count. Adverse effects from allopurinol can range from skin rashes to life-threatening severe allopurinol hypersensitivity, especially in HLA-B*5801-positive patients.[1][100]
Allopurinol
Allopurinol is converted to its active metabolite oxypurinol in the liver and has a half-life of 24 hours. The initial allopurinol dose is 100 mg daily in patients with CrCl more significant than 60 mL/min and is titrated upward by 100 mg every 2 to 4 weeks. A daily dose of 300 mg of allopurinol reduces serum urate levels in 33% of the population. Allopurinol can be increased above 300 mg daily to achieve the target serum uric acid.
Allopurinol is taken once daily. Medications like allopurinol and oxypurinol lower the serum urate by a dual action of inhibiting xanthine oxidase inhibitor and by competing with phosphoribosylpyrophosphate in the salvage pathway and through suppressive effects of drug nucleotides on aminotransferase activity. Allopurinol also nonselectively inhibits pyrimidine metabolism. In patients with stage 3 or greater CKD, the starting dose of allopurinol should be 50 mg daily.[100]
Adverse effects associated with allopurinol include the potential to trigger gout flares, pruritic and maculopapular rashes, leukopenia, thrombocytopenia, diarrhea, and severe cutaneous adverse reactions. Bone marrow suppression is uncommon but may occur at very high doses or in patients with CKD. Allopurinol can lead to a drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, a life-threatening reaction to allopurinol.
Major hypersensitivity reactions like Steven-Johnson syndrome or toxic epidermal necrolysis may occur in major allopurinol hypersensitivity syndrome (AHS). The highest risk for AHS occurs in the first 60 days after initiating allopurinol therapy. Patients who carry the HLA-B*5801 allele are at increased risk for developing severe hypersensitivity reactions, which are more common in people of Han Chinese, Korean, or Thai descent.[100][134] Testing for this allele is advisable in high-risk patients. Starting at a low dose and gradually increasing it can decrease the risk of adverse reactions. The recommended starting dose is 1.5 mg per unit of estimated GFR.[135] Interestingly, allopurinol can be safely increased above 300 mg daily, even in patients with CKD, to achieve the target serum uric acid.[136](B2)
Allopurinol can enhance the cytolytic and immunosuppressive effects of azathioprine and 6-mercaptopurine (6-MP), as these drugs are partially metabolized by xanthine oxidase.[137] Therefore, allopurinol should be avoided in patients undergoing treatment with these agents.[138] Additionally, in patients on warfarin, their anticoagulation status must be carefully monitored when allopurinol is prescribed.(B2)
Febuxostat
Febuxostat is a selective XOI that occupies the access channel to the molybdenum-pterin active site of the enzyme. Renal elimination plays a minor role in febuxostat pharmacokinetics. FDA approval for febuxostat in treating patients with gout and hyperuricemia includes initial daily doses of 40 mg. If the urate levels do not normalize within 2 weeks, the dosage is increased to 80 mg daily. Studies have demonstrated the superior effectiveness of febuxostat over allopurinol (maximum dose of 300 mg daily).[139][140] However, febuxostat may be more common with allopurinol than cardiovascular and hepatic abnormalities. In patients with CKD, febuxostat exhibits a more potent urate-lowering than allopurinol. Febuxostat has a distinct chemical structure, making it an option for patients who have experienced hypersensitivity reactions to allopurinol. Patients taking azathioprine, 6-MP, and theophylline are considered contraindications for the use of febuxostat.(A1)
In the CARES trial, which focused on cardiovascular safety in patients with gout and a history of cardiovascular disease, febuxostat and allopurinol were compared.[141] The primary endpoint, a composite of cardiovascular death, nonfatal MI, nonfatal stroke, or unstable angina requiring revascularization, showed no significant difference between the 2 drugs. However, febuxostat was associated with an increased risk of cardiovascular death (HR of 1.34, 95% CI 1.03-1.73, P=0.03) and higher all-cause mortality (HR of 1.22, 95% CI 1.01-1.47, P=0.04). Some population studies have also shown an increased risk of cardiovascular events and death.[142][143] However, some studies do not show an increased risk of cardiovascular events, including a randomized, open-label noninferiority study,[144] 2 population studies,[145][146] and a systematic review[147]. In a follow-up investigation of the CARES trial, patients who discontinued ULT experienced increased cardiovascular events and deaths at 30 days and 6 months.[148](A1)
Allopurinol and febuxostat are similarly effective, although some data suggest that febuxostat may be more effective in patients with CKD. In a comparative noninferiority trial of allopurinol and febuxostat, where at least 33% of patients had stage 3 CKD, both drugs showed similar efficacy in managing flares and reducing serum uric acid levels to the target range.[149](B2)
XOIs have demonstrated various effects, particularly in population studies focusing on cardiovascular disease.[150] The theory is that chronic hyperuricemia and MSU deposition result in chronic inflammation, thereby enhancing the progression of atherosclerosis. Notably, allopurinol has been associated with a modest reduction in all-cause mortality among patients with gout.[151][152] A case-matched cohort study conducted in Taiwan revealed that patients with gout faced an increased risk of cardiovascular and all-cause mortality. However, ULT treatment was linked to a reduced risk of cardiovascular (HR 0.29, 95% CI 0.11-0.80) and all-cause mortality (HR 0.47, 95% CI 0.29-0.79).[153] Allopurinol use was correlated with a lower risk of developing incident atrial fibrillation.[154] (B2)
ULT may also slow the progression of CKD,[155][156][157][158] and allopurinol is associated with a lower risk of incident renal disease in elderly patients compared to febuxostat.[159] Literature suggests that ULT in gout patients might affect outcomes, including dementia, erectile dysfunction, and other comorbidities. While some controlled trials have explored the effect of allopurinol on the incident rate of cardiovascular events, renal disease, and DM, these studies were performed in at-risk patients, not specifically in those with gout. Therefore, the relevance of these findings to patients with gout remains unclear. (B2)
Uricosuric Drugs
The uricosuric agents work by increasing renal urate clearance.[1][66] Patients with low or normal urinary uric acid excretion in the presence of hyperuricemia are potential candidates for uricosuric therapy. Drugs in this class include probenecid and lesinurad (withdrawn from the US market). These agents inhibit URAT1 at the apical membrane of the renal proximal tubule epithelial cell. However, they are ineffective as monotherapy in patients with low creatinine clearance (<30 ml/min) and contraindicated in patients with a history of nephrolithiasis.[160]
Probenecid, the only agent approved as a monotherapy, is initiated at 250 mg twice daily. Dose adjustments are made according to the serum urate concentration level, with increments every few weeks. The usual maintenance dose ranges from 500 to 1000 mg (taken 2 to 3 times daily), aiming to achieve target urate levels of less than 6 mg/dL (<357 µmol/L).
The significant side effects of uricosuric drugs are the precipitation of a gout flare, uric acid urolithiasis, gastrointestinal intolerance, and rash. Uricosuric agents are not appropriate for patients with CKD and a creatinine clearance of less than 60 mL/min. Patients with tophi are best treated with XOIs or pegloticase.
Uricase Pegloticase (urate oxidase)
Uricase is present in nonprimates and lower primates. Pegloticase, a pegylated recombinant form of uricase, is a potent agent that rapidly reduces serum urate levels by directly degrading uric acid into highly soluble allantoin. Polyethylene glycol (PEG) molecules are attached to the recombinant porcine-baboon uricase in a process known as PEGylation.This process extends the PEG molecule's half-life to days or weeks and decreases but does not eliminate immunogenicity.[161] (B3)
Pegloticase is reserved for patients with refractory gout, usually those with a high tophaceous burden. Patients must discontinue ULT while starting this medication because antibodies against pegloticase may develop. Pegloticase is administered as intravenous infusions every 2 weeks, and before each infusion, serum urate levels should be monitored to confirm urate-lowering efficacy. If the serum uric acid rises above 4 mg/dL, the infusions should be stopped, indicating that the patient is developing antibodies to pegloticase, which could lead to infusion reactions.
Pegloticase has effectively lowered serum uric acid in patients with refractory gout, as evidenced by short and long-term clinical trials.[162][163] Phase 3 studies revealed complete resolution of 1 or more tophi in 20% of patients by 13 weeks and lowered uric acid levels to less than 6 mg/dl in 42% of subjects within 6 months.[164] During the initial 6 months of pegloticase therapy, all patients should receive gout flare prophylaxis.(A1)
Due to the risk of severe hemolytic anemia, pegloticase is contraindicated in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. Acute gout flares are observed in 80% of patients during the first few months of pegloticase therapy, even with prophylactic measures in place. Moderate infusion reactions like flushing, urticaria, and hypotension are expected, with 2% of patients experiencing severe reactions like anaphylaxis. Some reactions, such as severe muscle pain and cramping, occur due to unknown mechanisms. Efforts to reduce the immunogenicity of pegloticase with concomitant use of methotrexate or mycophenolate mofetil have been effective, although infusion reactions remain a major issue.
Rasburicase, a nonpegylated recombinant uricase, has not received FDA approval for gout treatment. It prevents acute uric acid nephropathy due to tumor lysis syndrome in patients with high-risk leukemia and lymphoma.
Other Drugs With an Effect on Serum Uric Acid
Several drugs used to treat conditions like hypertension, type 2 DM, and HLD can affect the serum uric acid (see Table. Urate-Lowering Drugs and Mechanisms and Table. Urate-Increasing Drugs and Mechanisms).[165] The sodium-glucose cotransporter-2 inhibitors (SGLT2i) are particularly noteworthy. Studies have demonstrated their effectiveness in lowering serum uric acid levels.[166] In an investigation on the effect of empagliflozin therapy on heart failure, significant interactions were observed between empagliflozin treatment and baseline serum uric acid levels, affecting cardiovascular and all-cause mortality.[167] Additionally, SGLT2 inhibitors have been shown to reduce the risk of developing incident gout and acute flares of gouty arthritis.[167][168] (A1)
Table 8. Urate-Lowering Drugs and Mechanisms [165]
| Drug class | Drug | Mechanism |
| Antihypertensive |
Losartan CCBs |
Increases excretion, decreases URAT1 Various |
|
Anti-inflammatory Immunosuppressive |
High dose aspirin Leflunomide |
Biphasic effect on resorption Increases excretion |
| Lipid-lowering |
Statins Fenofibrate |
Unknown Increases excretion |
| Metabolism modulator | SGLT2 inhibitors | Increases excretion, GLUT9 |
| Sex hormone | Estrogen | Decreases resorption |
Table 9. Urate-Increasing Drugs and Mechanisms [165]
| Drug class | Drug | Mechanism |
| Diuretic | Loop diuretics |
Decreases excretion, decreases MRP4 Increases resorption, increases URAT1 |
| Other antihypertensive |
Thiazide diuretics
Beta-blockers |
Decreases excretion, decreases MRP4 Increases resorption, increases URAT1 Unknown |
| Antituberculosis drug |
Pyrazinamide Ethambutol |
Increase resorption, increases URAT1 Decreased renal clearance |
|
Anti-inflammatory Immunosuppressive |
Low-dose aspirin Calcineurin inhibitors |
Biphasic effect on resportion Decreases renal clearance |
| Metabolism modulator |
Lactate Insulin |
Increases resorption, increases URAT1 Increases resorption, increases URAT1 |
| Sex hormone | Testosterone | Increases resorption, increases URAT1 |
Differential Diagnosis
The differential diagnosis for an acute gout flare includes the following:
- Calcium pyrophosphate crystal deposition disease
- Basic calcium phosphate crystal disease
- Septic arthritis (crystal arthritis and septic arthritis may coexist) [92]
- OA
- Psoriatic arthritis
- Cellulitis
- Trauma
The differential diagnosis for tophaceous gout includes:
- Dactylitis
- Rheumatoid arthritis
- Osteomyelitis
Prognosis
The prognosis of gout varies based on the individual comorbidities. Patients with cardiovascular comorbidities tend to have higher mortality rates. Most patients can lead an everyday life with mild sequelae with proper treatment. Those experiencing symptoms early in life often present with more severe disease. Without lifestyle modifications, recurrent flare-ups are common.
Complications
Complications of gout are diverse and may encompass various systemic issues, including the following:[169]
Skeletal Complications
- Tophi
- Joint deformity
- OA
- Bone loss
Urological Complications
- Urate nephropathy
- Nephrolithiasis
Ocular Complications
- Conjunctivitis
- Uveitis
- Scleritis
Deterrence and Patient Education
Patients should be educated about lifestyle modifications and strategies to reduce the risk of gout flares and the condition's progression. Important points to discuss with patients include:
- Lifestyle changes are encouraged for patients with gout, including weight loss, limiting alcohol consumption, and avoiding certain foods. While these changes can significantly complement medical therapy, they might not always suffice to manage or reverse gout effectively.
- Weight gain and increased adiposity are risk factors for gout. In individuals with established gout who are overweight, weight loss is likely beneficial, leading to reductions in serum urate and alleviation of gout symptoms.[170][171]
- The optimal diet composition for managing gout includes adequate protein intake, especially from plant sources and low-fat dairy sources, while reducing consumption of animal sources high in purine, such as shellfish or red meat. Decreasing saturated fat intake and replacing simple sugars with complex carbohydrates is essential.
- It is advisable to avoid or significantly reduce the consumption of sugar-sweetened juices, alcoholic beverages, and drinks containing high-fructose corn syrup.
Pearls and Other Issues
IL-1 is an important mediator of inflammation in gout and represents a potential therapeutic target for managing gout flares.[172] For patients with multiple medical comorbidities or are on anticoagulation, a short-acting IL-1 inhibitor, like anakinra, can be considered an an alternative treatment for gout flare as an alternative to the first-line therapies.
Enhancing Healthcare Team Outcomes
Most patients with gout commonly have accompanying comorbidities. The prevalence of gout is higher among individuals with chronic diseases such as HTN, CKD, DM, obesity, CHF, and MI.[173]
Gout treatment necessitates a collaborative approach from an entire interprofessional healthcare team. The healthcare professional must swiftly identify the pathology and rule out other causes. In some cases, a rheumatology consult might be necessary. When developing pharmacological approaches, considering comorbidities is essential, and monitoring their response to treatment is crucial.
The pharmacist and nurse are pivotal in educating the patient on medication compliance. Pharmacists should also assist the team by conducting medication reconciliation, verifying appropriate dosing, and providing input on agent selection if the initial treatment proves ineffective.
Dietitians should encourage patients to abstain from alcohol, avoid purine-rich foods, and maintain a healthy body weight. The collaborative efforts of specialists, primary care providers, nurses, nurse practitioners, and dieticians are instrumental in reducing gout morbidity associated with gout. Effective communication among all team members is vital in coordinating patient education on lifestyle modifications, significantly reducing the risk and frequency of gout flare-ups.
Healthcare providers, including primary care physicians, physician assistants, and nurse practitioners, should be adept at identifying classic gout symptoms and have a low threshold for referring patients for an arthrocentesis if there is any uncertainty about the diagnosis. Collaborating with the interprofessional team, as detailed earlier, is essential.
Referral to a specialist, such as a rheumatologist, should be considered for patients with joint pain in the following situations:
- Unclear etiology with hyperuricemia
- Unclear etiology with normal serum urate level
- Patients with renal impairment
- Failed trial of XOI treatment
- Multiple side effects from the medications
- Refractory gout [102]
Only through an interprofessional team approach with close communication can the morbidity of gout be lowered. Directing the treatment appropriately and maintaining active communication within the team is vital to achieving favorable outcomes.
Media
(Click Image to Enlarge)
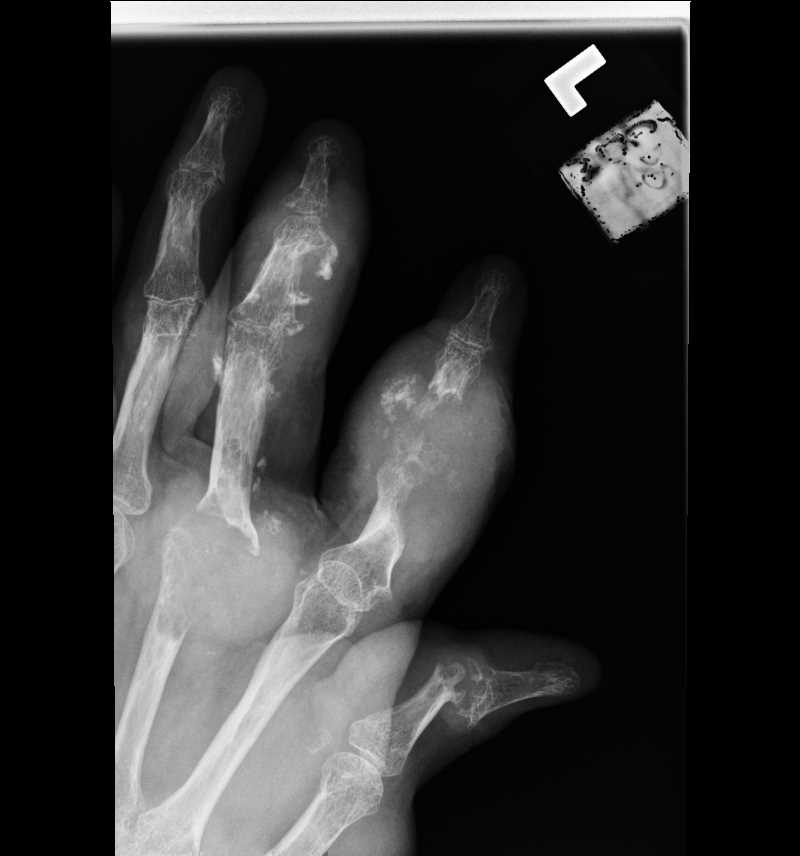
Hand Radiograph, Gout
Contributed by Scott Dulebohn, MD
(Click Image to Enlarge)
![Gout, [SATA]](/pictures/getimagecontent//6491)
Gout, [SATA]
Contributed by Steve Bhmji, MS, MD, PhD
(Click Image to Enlarge)
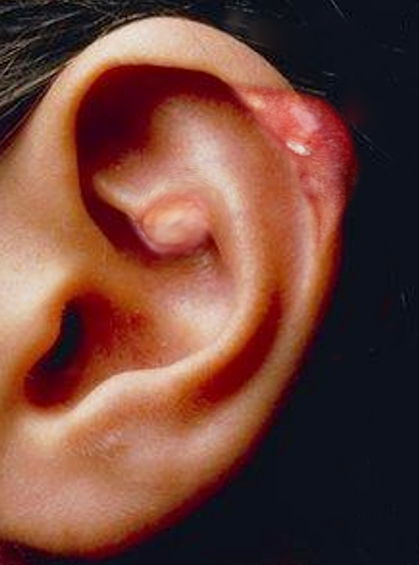
Gout in the Ear
Image courtesy S Bhimji MD
(Click Image to Enlarge)
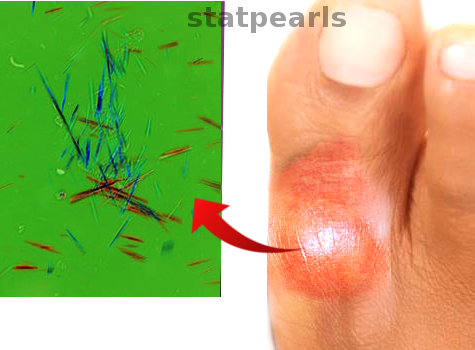
Acute gout attack
Image courtesy O.Chaigasame
(Click Image to Enlarge)
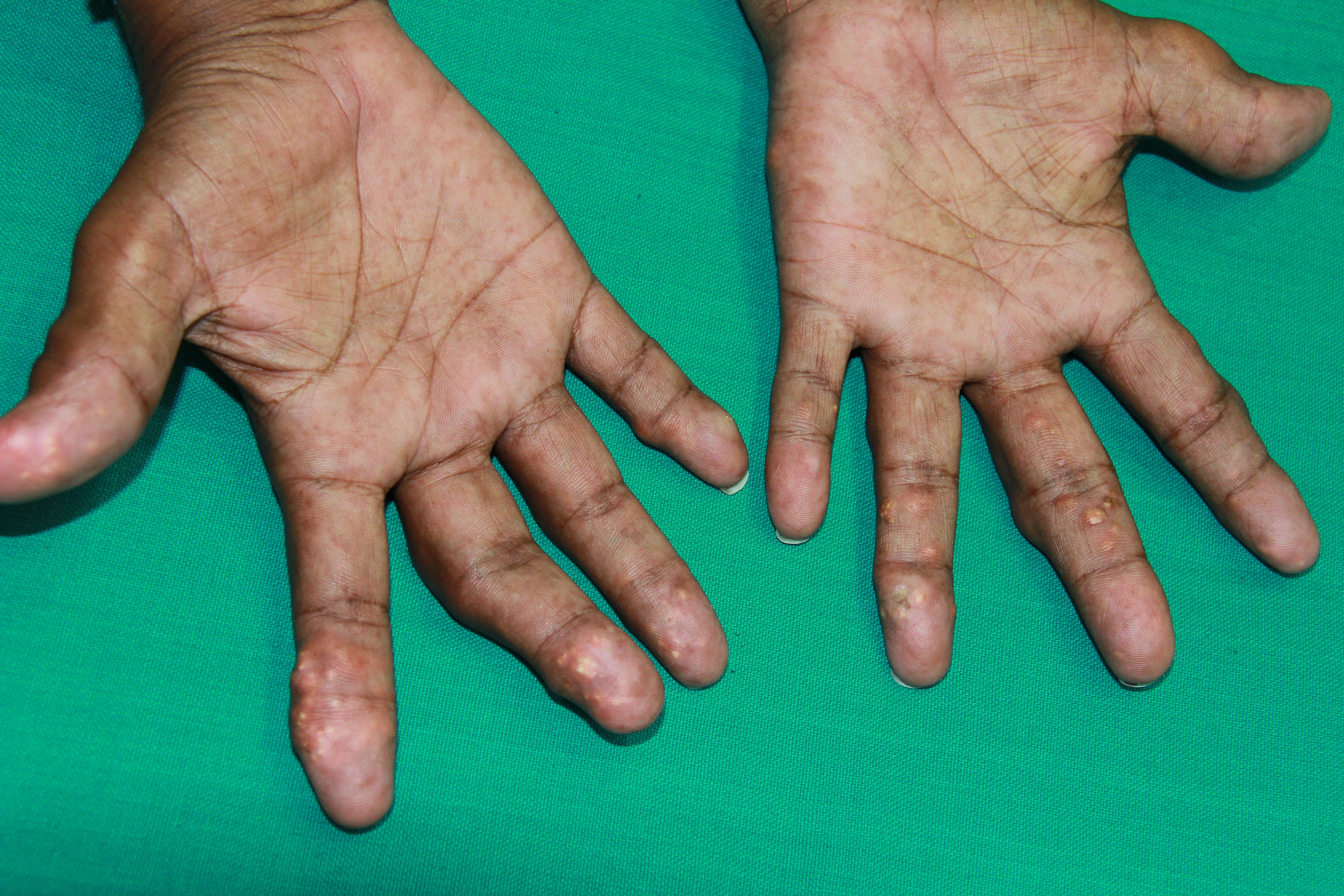
Gout Tophi
Contributed by Dr. Shyam Verma, MBBS, DVD, FRCP, FAAD, Vadodara, India
References
Neogi T. Gout. Annals of internal medicine. 2016 Jul 5:165(1):ITC1-ITC16. doi: 10.7326/AITC201607050. Epub [PubMed PMID: 27380294]
Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet (London, England). 2016 Oct 22:388(10055):2039-2052. doi: 10.1016/S0140-6736(16)00346-9. Epub 2016 Apr 21 [PubMed PMID: 27112094]
Richette P, Bardin T. Gout. Lancet (London, England). 2010 Jan 23:375(9711):318-28. doi: 10.1016/S0140-6736(09)60883-7. Epub 2009 Aug 17 [PubMed PMID: 19692116]
Nuki G, Simkin PA. A concise history of gout and hyperuricemia and their treatment. Arthritis research & therapy. 2006:8 Suppl 1(Suppl 1):S1 [PubMed PMID: 16820040]
Loeb JN. The influence of temperature on the solubility of monosodium urate. Arthritis and rheumatism. 1972 Mar-Apr:15(2):189-92 [PubMed PMID: 5027604]
Nian YL, You CG. Susceptibility genes of hyperuricemia and gout. Hereditas. 2022 Aug 4:159(1):30. doi: 10.1186/s41065-022-00243-y. Epub 2022 Aug 4 [PubMed PMID: 35922835]
Merriman TR, Choi HK, Dalbeth N. The genetic basis of gout. Rheumatic diseases clinics of North America. 2014 May:40(2):279-90. doi: 10.1016/j.rdc.2014.01.009. Epub 2014 Feb 19 [PubMed PMID: 24703347]
Choi HK, Mount DB, Reginato AM, American College of Physicians, American Physiological Society. Pathogenesis of gout. Annals of internal medicine. 2005 Oct 4:143(7):499-516 [PubMed PMID: 16204163]
Tan PK, Farrar JE, Gaucher EA, Miner JN. Coevolution of URAT1 and Uricase during Primate Evolution: Implications for Serum Urate Homeostasis and Gout. Molecular biology and evolution. 2016 Sep:33(9):2193-200. doi: 10.1093/molbev/msw116. Epub 2016 Jun 26 [PubMed PMID: 27352852]
Level 3 (low-level) evidenceAbhishek A, Roddy E, Doherty M. Gout - a guide for the general and acute physicians. Clinical medicine (London, England). 2017 Feb:17(1):54-59. doi: 10.7861/clinmedicine.17-1-54. Epub [PubMed PMID: 28148582]
Scott JT. Asymptomatic hyperuricaemia. British medical journal (Clinical research ed.). 1987 Apr 18:294(6578):987-8 [PubMed PMID: 3119013]
Dalbeth N, Phipps-Green A, Frampton C, Neogi T, Taylor WJ, Merriman TR. Relationship between serum urate concentration and clinically evident incident gout: an individual participant data analysis. Annals of the rheumatic diseases. 2018 Jul:77(7):1048-1052. doi: 10.1136/annrheumdis-2017-212288. Epub 2018 Feb 20 [PubMed PMID: 29463518]
Wu EQ, Patel PA, Mody RR, Yu AP, Cahill KE, Tang J, Krishnan E. Frequency, risk, and cost of gout-related episodes among the elderly: does serum uric acid level matter? The Journal of rheumatology. 2009 May:36(5):1032-40. doi: 10.3899/jrheum.080487. Epub 2009 Apr 15 [PubMed PMID: 19369467]
Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ (Clinical research ed.). 2008 Feb 9:336(7639):309-12. doi: 10.1136/bmj.39449.819271.BE. Epub 2008 Jan 31 [PubMed PMID: 18244959]
Level 2 (mid-level) evidenceChoi HK, Willett W, Curhan G. Fructose-rich beverages and risk of gout in women. JAMA. 2010 Nov 24:304(20):2270-8. doi: 10.1001/jama.2010.1638. Epub 2010 Nov 10 [PubMed PMID: 21068145]
Dalbeth N, So A. Hyperuricaemia and gout: state of the art and future perspectives. Annals of the rheumatic diseases. 2010 Oct:69(10):1738-43. doi: 10.1136/ard.2010.136218. Epub [PubMed PMID: 20858623]
Level 3 (low-level) evidenceChoi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. The New England journal of medicine. 2004 Mar 11:350(11):1093-103 [PubMed PMID: 15014182]
Juraschek SP, Yokose C, McCormick N, Miller ER 3rd, Appel LJ, Choi HK. Effects of Dietary Patterns on Serum Urate: Results From a Randomized Trial of the Effects of Diet on Hypertension. Arthritis & rheumatology (Hoboken, N.J.). 2021 Jun:73(6):1014-1020. doi: 10.1002/art.41614. Epub 2021 Apr 23 [PubMed PMID: 33615722]
Level 1 (high-level) evidenceRai SK, Fung TT, Lu N, Keller SF, Curhan GC, Choi HK. The Dietary Approaches to Stop Hypertension (DASH) diet, Western diet, and risk of gout in men: prospective cohort study. BMJ (Clinical research ed.). 2017 May 9:357():j1794. doi: 10.1136/bmj.j1794. Epub 2017 May 9 [PubMed PMID: 28487277]
Huang HY, Appel LJ, Choi MJ, Gelber AC, Charleston J, Norkus EP, Miller ER 3rd. The effects of vitamin C supplementation on serum concentrations of uric acid: results of a randomized controlled trial. Arthritis and rheumatism. 2005 Jun:52(6):1843-7 [PubMed PMID: 15934094]
Level 1 (high-level) evidenceGao X, Curhan G, Forman JP, Ascherio A, Choi HK. Vitamin C intake and serum uric acid concentration in men. The Journal of rheumatology. 2008 Sep:35(9):1853-8 [PubMed PMID: 18464304]
Juraschek SP, Miller ER 3rd, Gelber AC. Effect of oral vitamin C supplementation on serum uric acid: a meta-analysis of randomized controlled trials. Arthritis care & research. 2011 Sep:63(9):1295-306. doi: 10.1002/acr.20519. Epub [PubMed PMID: 21671418]
Level 1 (high-level) evidenceChoi HK, Gao X, Curhan G. Vitamin C intake and the risk of gout in men: a prospective study. Archives of internal medicine. 2009 Mar 9:169(5):502-7. doi: 10.1001/archinternmed.2008.606. Epub [PubMed PMID: 19273781]
Juraschek SP, Gaziano JM, Glynn RJ, Gomelskaya N, Bubes VY, Buring JE, Shmerling RH, Sesso HD. Effects of vitamin C supplementation on gout risk: results from the Physicians' Health Study II trial. The American journal of clinical nutrition. 2022 Sep 2:116(3):812-819. doi: 10.1093/ajcn/nqac140. Epub [PubMed PMID: 35575611]
Jacob RA, Spinozzi GM, Simon VA, Kelley DS, Prior RL, Hess-Pierce B, Kader AA. Consumption of cherries lowers plasma urate in healthy women. The Journal of nutrition. 2003 Jun:133(6):1826-9 [PubMed PMID: 12771324]
Zhang Y, Neogi T, Chen C, Chaisson C, Hunter DJ, Choi HK. Cherry consumption and decreased risk of recurrent gout attacks. Arthritis and rheumatism. 2012 Dec:64(12):4004-11. doi: 10.1002/art.34677. Epub [PubMed PMID: 23023818]
Glynn RJ, Campion EW, Silbert JE. Trends in serum uric acid levels 1961--1980. Arthritis and rheumatism. 1983 Jan:26(1):87-93 [PubMed PMID: 6824508]
Takiue Y, Hosoyamada M, Kimura M, Saito H. The effect of female hormones upon urate transport systems in the mouse kidney. Nucleosides, nucleotides & nucleic acids. 2011 Feb:30(2):113-9. doi: 10.1080/15257770.2010.551645. Epub [PubMed PMID: 21360409]
Level 3 (low-level) evidenceRho YH, Zhu Y, Choi HK. The epidemiology of uric acid and fructose. Seminars in nephrology. 2011 Sep:31(5):410-9. doi: 10.1016/j.semnephrol.2011.08.004. Epub [PubMed PMID: 22000647]
Hak AE, Choi HK. Menopause, postmenopausal hormone use and serum uric acid levels in US women--the Third National Health and Nutrition Examination Survey. Arthritis research & therapy. 2008:10(5):R116. doi: 10.1186/ar2519. Epub 2008 Sep 26 [PubMed PMID: 18822120]
Level 3 (low-level) evidenceArromdee E, Michet CJ, Crowson CS, O'Fallon WM, Gabriel SE. Epidemiology of gout: is the incidence rising? The Journal of rheumatology. 2002 Nov:29(11):2403-6 [PubMed PMID: 12415600]
Level 2 (mid-level) evidenceChoi H. Epidemiology of crystal arthropathy. Rheumatic diseases clinics of North America. 2006 May:32(2):255-73, v [PubMed PMID: 16716879]
Ragab G, Elshahaly M, Bardin T. Gout: An old disease in new perspective - A review. Journal of advanced research. 2017 Sep:8(5):495-511. doi: 10.1016/j.jare.2017.04.008. Epub 2017 May 10 [PubMed PMID: 28748116]
Level 3 (low-level) evidenceLawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, Gabriel S, Hirsch R, Hochberg MC, Hunder GG, Jordan JM, Katz JN, Kremers HM, Wolfe F, National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis and rheumatism. 2008 Jan:58(1):26-35. doi: 10.1002/art.23176. Epub [PubMed PMID: 18163497]
Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis and rheumatism. 2011 Oct:63(10):3136-41. doi: 10.1002/art.30520. Epub [PubMed PMID: 21800283]
Level 3 (low-level) evidenceKramer HM, Curhan G. The association between gout and nephrolithiasis: the National Health and Nutrition Examination Survey III, 1988-1994. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2002 Jul:40(1):37-42 [PubMed PMID: 12087559]
Level 3 (low-level) evidenceMcCormick N, Lu N, Yokose C, Joshi AD, Sheehy S, Rosenberg L, Warner ET, Dalbeth N, Merriman TR, Saag KG, Zhang Y, Choi HK. Racial and Sex Disparities in Gout Prevalence Among US Adults. JAMA network open. 2022 Aug 1:5(8):e2226804. doi: 10.1001/jamanetworkopen.2022.26804. Epub 2022 Aug 1 [PubMed PMID: 35969396]
Elfishawi MM, Zleik N, Kvrgic Z, Michet CJ Jr, Crowson CS, Matteson EL, Bongartz T. The Rising Incidence of Gout and the Increasing Burden of Comorbidities: A Population-based Study over 20 Years. The Journal of rheumatology. 2018 Apr:45(4):574-579. doi: 10.3899/jrheum.170806. Epub 2017 Dec 15 [PubMed PMID: 29247151]
Kuo CF, Grainge MJ, Mallen C, Zhang W, Doherty M. Rising burden of gout in the UK but continuing suboptimal management: a nationwide population study. Annals of the rheumatic diseases. 2015 Apr:74(4):661-7. doi: 10.1136/annrheumdis-2013-204463. Epub 2014 Jan 15 [PubMed PMID: 24431399]
Level 2 (mid-level) evidenceBai L, Zhou JB, Zhou T, Newson RB, Cardoso MA. Incident gout and weight change patterns: a retrospective cohort study of US adults. Arthritis research & therapy. 2021 Mar 2:23(1):69. doi: 10.1186/s13075-021-02461-7. Epub 2021 Mar 2 [PubMed PMID: 33653403]
Level 2 (mid-level) evidenceHwang J, Lee MY, Ahn JK, Cha HS. Relationship Between Changing Body Mass Index and Serum Uric Acid Alteration Among Clinically Apparently Healthy Korean Men. Arthritis care & research. 2022 Aug:74(8):1277-1286. doi: 10.1002/acr.24576. Epub 2022 Apr 30 [PubMed PMID: 33544980]
Liu K, Yao Y, Chen W, Mao Y, Ye D, Wen C. Modifiable risk factors and incidence of gout: Estimation of population attributable fraction in the US. Seminars in arthritis and rheumatism. 2022 Aug:55():152040. doi: 10.1016/j.semarthrit.2022.152040. Epub 2022 Jun 3 [PubMed PMID: 35679791]
Annemans L, Spaepen E, Gaskin M, Bonnemaire M, Malier V, Gilbert T, Nuki G. Gout in the UK and Germany: prevalence, comorbidities and management in general practice 2000-2005. Annals of the rheumatic diseases. 2008 Jul:67(7):960-6 [PubMed PMID: 17981913]
Level 2 (mid-level) evidenceKuo CF, Grainge MJ, Mallen C, Zhang W, Doherty M. Comorbidities in patients with gout prior to and following diagnosis: case-control study. Annals of the rheumatic diseases. 2016 Jan:75(1):210-7. doi: 10.1136/annrheumdis-2014-206410. Epub 2014 Nov 14 [PubMed PMID: 25398375]
Level 2 (mid-level) evidenceKrishnan E. Chronic kidney disease and the risk of incident gout among middle-aged men: a seven-year prospective observational study. Arthritis and rheumatism. 2013 Dec:65(12):3271-8. doi: 10.1002/art.38171. Epub [PubMed PMID: 23982888]
Level 2 (mid-level) evidenceMoon KW, Kim MJ, Choi IA, Shin K. Cardiovascular Risks in Korean Patients with Gout: Analysis Using a National Health Insurance Service Database. Journal of clinical medicine. 2022 Apr 11:11(8):. doi: 10.3390/jcm11082124. Epub 2022 Apr 11 [PubMed PMID: 35456221]
Cipolletta E, Tata LJ, Nakafero G, Avery AJ, Mamas MA, Abhishek A. Association Between Gout Flare and Subsequent Cardiovascular Events Among Patients With Gout. JAMA. 2022 Aug 2:328(5):440-450. doi: 10.1001/jama.2022.11390. Epub [PubMed PMID: 35916846]
Disveld IJM, Zoakman S, Jansen TLTA, Rongen GA, Kienhorst LBE, Janssens HJEM, Fransen J, Janssen M. Crystal-proven gout patients have an increased mortality due to cardiovascular diseases, cancer, and infectious diseases especially when having tophi and/or high serum uric acid levels: a prospective cohort study. Clinical rheumatology. 2019 May:38(5):1385-1391. doi: 10.1007/s10067-019-04520-6. Epub 2019 Mar 30 [PubMed PMID: 30929152]
Ioachimescu AG, Brennan DM, Hoar BM, Hazen SL, Hoogwerf BJ. Serum uric acid is an independent predictor of all-cause mortality in patients at high risk of cardiovascular disease: a preventive cardiology information system (PreCIS) database cohort study. Arthritis and rheumatism. 2008 Feb:58(2):623-30. doi: 10.1002/art.23121. Epub [PubMed PMID: 18240236]
Level 2 (mid-level) evidenceVargas-Santos AB, Neogi T, da Rocha Castelar-Pinheiro G, Kapetanovic MC, Turkiewicz A. Cause-Specific Mortality in Gout: Novel Findings of Elevated Risk of Non-Cardiovascular-Related Deaths. Arthritis & rheumatology (Hoboken, N.J.). 2019 Nov:71(11):1935-1942. doi: 10.1002/art.41008. Epub 2019 Sep 25 [PubMed PMID: 31169353]
Hong JY, Lan TY, Tang GJ, Tang CH, Chen TJ, Lin HY. Gout and the risk of dementia: a nationwide population-based cohort study. Arthritis research & therapy. 2015 May 28:17(1):139. doi: 10.1186/s13075-015-0642-1. Epub 2015 May 28 [PubMed PMID: 26018424]
Min KH, Kang SO, Oh SJ, Han JM, Lee KE. Association Between Gout and Dementia in the Elderly: A Nationwide Population-Based Cohort Study. The American journal of geriatric psychiatry : official journal of the American Association for Geriatric Psychiatry. 2021 Dec:29(12):1177-1185. doi: 10.1016/j.jagp.2021.01.016. Epub 2021 Jan 30 [PubMed PMID: 33593591]
Pan SY, Cheng RJ, Xia ZJ, Zhang QP, Liu Y. Risk of dementia in gout and hyperuricaemia: a meta-analysis of cohort studies. BMJ open. 2021 Jun 22:11(6):e041680. doi: 10.1136/bmjopen-2020-041680. Epub 2021 Jun 22 [PubMed PMID: 34158290]
Level 1 (high-level) evidenceKim JH, Yim DH, Choi IA, Lee J, Park H, Eom SY. Impact of Clinical Association Between Gout and Dementia: A Nationwide Population-Based Cohort Study in Korea. Arthritis care & research. 2023 May:75(5):1088-1094. doi: 10.1002/acr.24959. Epub 2022 Dec 18 [PubMed PMID: 35604886]
Lu N, Dubreuil M, Zhang Y, Neogi T, Rai SK, Ascherio A, Hernán MA, Choi HK. Gout and the risk of Alzheimer's disease: a population-based, BMI-matched cohort study. Annals of the rheumatic diseases. 2016 Mar:75(3):547-51. doi: 10.1136/annrheumdis-2014-206917. Epub 2015 Mar 4 [PubMed PMID: 25739830]
Level 2 (mid-level) evidenceSingh JA, Cleveland JD. Gout and dementia in the elderly: a cohort study of Medicare claims. BMC geriatrics. 2018 Nov 14:18(1):281. doi: 10.1186/s12877-018-0975-0. Epub 2018 Nov 14 [PubMed PMID: 30428833]
Latourte A, Soumaré A, Bardin T, Perez-Ruiz F, Debette S, Richette P. Uric acid and incident dementia over 12 years of follow-up: a population-based cohort study. Annals of the rheumatic diseases. 2018 Mar:77(3):328-335. doi: 10.1136/annrheumdis-2016-210767. Epub 2017 Jul 28 [PubMed PMID: 28754803]
Alonso A, Rodríguez LA, Logroscino G, Hernán MA. Gout and risk of Parkinson disease: a prospective study. Neurology. 2007 Oct 23:69(17):1696-700 [PubMed PMID: 17954784]
Level 2 (mid-level) evidenceDe Vera M, Rahman MM, Rankin J, Kopec J, Gao X, Choi H. Gout and the risk of Parkinson's disease: a cohort study. Arthritis and rheumatism. 2008 Nov 15:59(11):1549-54. doi: 10.1002/art.24193. Epub [PubMed PMID: 18975349]
Level 2 (mid-level) evidencePou MA, Orfila F, Pagonabarraga J, Ferrer-Moret S, Corominas H, Diaz-Torne C. Risk of Parkinson's disease in a gout Mediterranean population: A case-control study. Joint bone spine. 2022 Nov:89(6):105402. doi: 10.1016/j.jbspin.2022.105402. Epub 2022 Apr 30 [PubMed PMID: 35504516]
Level 2 (mid-level) evidenceFazlollahi A, Zahmatyar M, Alizadeh H, Noori M, Jafari N, Nejadghaderi SA, Sullman MJM, Gharagozli K, Kolahi AA, Safiri S. Association between gout and the development of Parkinson's disease: a systematic review and meta-analysis. BMC neurology. 2022 Oct 11:22(1):383. doi: 10.1186/s12883-022-02874-0. Epub 2022 Oct 11 [PubMed PMID: 36221048]
Level 1 (high-level) evidenceHu LY, Yang AC, Lee SC, You ZH, Tsai SJ, Hu CK, Shen CC. Risk of Parkinson's disease following gout: a population-based retrospective cohort study in Taiwan. BMC neurology. 2020 Sep 8:20(1):338. doi: 10.1186/s12883-020-01916-9. Epub 2020 Sep 8 [PubMed PMID: 32900384]
Level 2 (mid-level) evidenceSingh JA, Cleveland JD. Gout and the risk of Parkinson's disease in older adults: a study of U.S. Medicare data. BMC neurology. 2019 Jan 5:19(1):4. doi: 10.1186/s12883-018-1234-x. Epub 2019 Jan 5 [PubMed PMID: 30611222]
Bursill D, Taylor WJ, Terkeltaub R, Abhishek A, So AK, Vargas-Santos AB, Gaffo AL, Rosenthal A, Tausche AK, Reginato A, Manger B, Sciré C, Pineda C, van Durme C, Lin CT, Yin C, Albert DA, Biernat-Kaluza E, Roddy E, Pascual E, Becce F, Perez-Ruiz F, Sivera F, Lioté F, Schett G, Nuki G, Filippou G, McCarthy G, da Rocha Castelar Pinheiro G, Ea HK, Tupinambá HA, Yamanaka H, Choi HK, Mackay J, ODell JR, Vázquez Mellado J, Singh JA, Fitzgerald JD, Jacobsson LTH, Joosten L, Harrold LR, Stamp L, Andrés M, Gutierrez M, Kuwabara M, Dehlin M, Janssen M, Doherty M, Hershfield MS, Pillinger M, Edwards NL, Schlesinger N, Kumar N, Slot O, Ottaviani S, Richette P, MacMullan PA, Chapman PT, Lipsky PE, Robinson P, Khanna PP, Gancheva RN, Grainger R, Johnson RJ, Te Kampe R, Keenan RT, Tedeschi SK, Kim S, Choi SJ, Fields TR, Bardin T, Uhlig T, Jansen T, Merriman T, Pascart T, Neogi T, Klück V, Louthrenoo W, Dalbeth N. Gout, Hyperuricaemia and Crystal-Associated Disease Network (G-CAN) consensus statement regarding labels and definitions of disease states of gout. Annals of the rheumatic diseases. 2019 Nov:78(11):1592-1600. doi: 10.1136/annrheumdis-2019-215933. Epub 2019 Sep 9 [PubMed PMID: 31501138]
Level 3 (low-level) evidenceBursill D, Taylor WJ, Terkeltaub R, Kuwabara M, Merriman TR, Grainger R, Pineda C, Louthrenoo W, Edwards NL, Andrés M, Vargas-Santos AB, Roddy E, Pascart T, Lin CT, Perez-Ruiz F, Tedeschi SK, Kim SC, Harrold LR, McCarthy G, Kumar N, Chapman PT, Tausche AK, Vazquez-Mellado J, Gutierrez M, da Rocha Castelar-Pinheiro G, Richette P, Pascual E, Fisher MC, Burgos-Vargas R, Robinson PC, Singh JA, Jansen TL, Saag KG, Slot O, Uhlig T, Solomon DH, Keenan RT, Scire CA, Biernat-Kaluza E, Dehlin M, Nuki G, Schlesinger N, Janssen M, Stamp LK, Sivera F, Reginato AM, Jacobsson L, Lioté F, Ea HK, Rosenthal A, Bardin T, Choi HK, Hershfield MS, Czegley C, Choi SJ, Dalbeth N. Gout, Hyperuricemia, and Crystal-Associated Disease Network Consensus Statement Regarding Labels and Definitions for Disease Elements in Gout. Arthritis care & research. 2019 Mar:71(3):427-434. doi: 10.1002/acr.23607. Epub [PubMed PMID: 29799677]
Level 3 (low-level) evidenceDalbeth N, Gosling AL, Gaffo A, Abhishek A. Gout. Lancet (London, England). 2021 May 15:397(10287):1843-1855. doi: 10.1016/S0140-6736(21)00569-9. Epub 2021 Mar 30 [PubMed PMID: 33798500]
Desai J, Steiger S, Anders HJ. Molecular Pathophysiology of Gout. Trends in molecular medicine. 2017 Aug:23(8):756-768. doi: 10.1016/j.molmed.2017.06.005. Epub 2017 Jul 18 [PubMed PMID: 28732688]
Chhana A, Lee G, Dalbeth N. Factors influencing the crystallization of monosodium urate: a systematic literature review. BMC musculoskeletal disorders. 2015 Oct 14:16():296. doi: 10.1186/s12891-015-0762-4. Epub 2015 Oct 14 [PubMed PMID: 26467213]
Level 1 (high-level) evidencePascual E, Addadi L, Andrés M, Sivera F. Mechanisms of crystal formation in gout-a structural approach. Nature reviews. Rheumatology. 2015 Dec:11(12):725-30. doi: 10.1038/nrrheum.2015.125. Epub 2015 Sep 15 [PubMed PMID: 26369610]
Franklin BS, Mangan MS, Latz E. Crystal Formation in Inflammation. Annual review of immunology. 2016 May 20:34():173-202. doi: 10.1146/annurev-immunol-041015-055539. Epub 2016 Jan 11 [PubMed PMID: 26772211]
Ginsberg MH, Kozin F. Mechanisms of cellular interaction with monosodium urate crystals. IgG-dependent and IgG-independent platelet stimulation by urate crystals. Arthritis and rheumatism. 1978 Nov-Dec:21(8):896-903 [PubMed PMID: 737013]
Giclas PC, Ginsberg MH, Cooper NR. Immunoglobulin G independent activation of the classical complement pathway by monosodium urate crystals. The Journal of clinical investigation. 1979 Apr:63(4):759-64 [PubMed PMID: 438335]
Fields TR, Abramson SB, Weissmann G, Kaplan AP, Ghebrehiwet B. Activation of the alternative pathway of complement by monosodium urate crystals. Clinical immunology and immunopathology. 1983 Feb:26(2):249-57 [PubMed PMID: 6872344]
So AK, Martinon F. Inflammation in gout: mechanisms and therapeutic targets. Nature reviews. Rheumatology. 2017 Nov:13(11):639-647. doi: 10.1038/nrrheum.2017.155. Epub 2017 Sep 28 [PubMed PMID: 28959043]
Chen J, Wu M, Yang J, Wang J, Qiao Y, Li X. The Immunological Basis in the Pathogenesis of Gout. Iranian journal of immunology : IJI. 2017 Jun:14(2):90-98 [PubMed PMID: 28630380]
Level 2 (mid-level) evidenceLee JH, Kim HS, Lee JH, Yang G, Kim HJ. Natural Products as a Novel Therapeutic Strategy for NLRP3 Inflammasome-Mediated Gout. Frontiers in pharmacology. 2022:13():861399. doi: 10.3389/fphar.2022.861399. Epub 2022 Mar 16 [PubMed PMID: 35370689]
Chhana A, Dalbeth N. Structural joint damage in gout. Rheumatic diseases clinics of North America. 2014 May:40(2):291-309. doi: 10.1016/j.rdc.2014.01.006. Epub 2014 Feb 20 [PubMed PMID: 24703348]
Dalbeth N, Clark B, Gregory K, Gamble G, Sheehan T, Doyle A, McQueen FM. Mechanisms of bone erosion in gout: a quantitative analysis using plain radiography and computed tomography. Annals of the rheumatic diseases. 2009 Aug:68(8):1290-5. doi: 10.1136/ard.2008.094201. Epub 2008 Aug 15 [PubMed PMID: 18708415]
Dalbeth N, Smith T, Nicolson B, Clark B, Callon K, Naot D, Haskard DO, McQueen FM, Reid IR, Cornish J. Enhanced osteoclastogenesis in patients with tophaceous gout: urate crystals promote osteoclast development through interactions with stromal cells. Arthritis and rheumatism. 2008 Jun:58(6):1854-65. doi: 10.1002/art.23488. Epub [PubMed PMID: 18512794]
Level 3 (low-level) evidenceMCCARTY DJ, HOLLANDER JL. Identification of urate crystals in gouty synovial fluid. Annals of internal medicine. 1961 Mar:54():452-60 [PubMed PMID: 13773775]
Towiwat P, Chhana A, Dalbeth N. The anatomical pathology of gout: a systematic literature review. BMC musculoskeletal disorders. 2019 Apr 1:20(1):140. doi: 10.1186/s12891-019-2519-y. Epub 2019 Apr 1 [PubMed PMID: 30935368]
Level 1 (high-level) evidenceHadler NM, Franck WA, Bress NM, Robinson DR. Acute polyarticular gout. The American journal of medicine. 1974 May:56(5):715-9 [PubMed PMID: 4825606]
Komarla A, Schumacher R, Merkel PA. Spinal gout presenting as acute low back pain. Arthritis and rheumatism. 2013 Oct:65(10):2660. doi: 10.1002/art.38069. Epub [PubMed PMID: 23818092]
Level 3 (low-level) evidenceLumezanu E, Konatalapalli R, Weinstein A. Axial (spinal) gout. Current rheumatology reports. 2012 Apr:14(2):161-4. doi: 10.1007/s11926-012-0236-8. Epub [PubMed PMID: 22318623]
Yü TF. Secondary gout associated with myeloproliferative diseases. Arthritis and rheumatism. 1965 Oct:8(5):765-71 [PubMed PMID: 5216775]
Level 3 (low-level) evidenceLin HY, Rocher LL, McQuillan MA, Schmaltz S, Palella TD, Fox IH. Cyclosporine-induced hyperuricemia and gout. The New England journal of medicine. 1989 Aug 3:321(5):287-92 [PubMed PMID: 2664517]
Choi HK, Niu J, Neogi T, Chen CA, Chaisson C, Hunter D, Zhang Y. Nocturnal risk of gout attacks. Arthritis & rheumatology (Hoboken, N.J.). 2015 Feb:67(2):555-62. doi: 10.1002/art.38917. Epub [PubMed PMID: 25504842]
Level 2 (mid-level) evidenceRakieh C, Conaghan PG. Diagnosis and treatment of gout in primary care. The Practitioner. 2011 Dec:255(1746):17-20, 2-3 [PubMed PMID: 22272526]
Shah D, Mohan G, Flueckiger P, Corrigan F, Conn D. Polyarticular Gout Flare Masquerading as Sepsis. The American journal of medicine. 2015 Jul:128(7):e11-2. doi: 10.1016/j.amjmed.2014.12.025. Epub 2015 Jan 20 [PubMed PMID: 25614957]
Level 3 (low-level) evidencePascual E, Batlle-Gualda E, Martínez A, Rosas J, Vela P. Synovial fluid analysis for diagnosis of intercritical gout. Annals of internal medicine. 1999 Nov 16:131(10):756-9 [PubMed PMID: 10577299]
Level 2 (mid-level) evidenceCoakley G, Mathews C, Field M, Jones A, Kingsley G, Walker D, Phillips M, Bradish C, McLachlan A, Mohammed R, Weston V, British Society for Rheumatology Standards, Guidelines and Audit Working Group. BSR & BHPR, BOA, RCGP and BSAC guidelines for management of the hot swollen joint in adults. Rheumatology (Oxford, England). 2006 Aug:45(8):1039-41 [PubMed PMID: 16829534]
Yu KH, Luo SF, Liou LB, Wu YJ, Tsai WP, Chen JY, Ho HH. Concomitant septic and gouty arthritis--an analysis of 30 cases. Rheumatology (Oxford, England). 2003 Sep:42(9):1062-6 [PubMed PMID: 12730521]
Level 3 (low-level) evidenceSivera F, Andrés M, Quilis N. Gout: Diagnosis and treatment. Medicina clinica. 2017 Mar 22:148(6):271-276. doi: 10.1016/j.medcli.2016.10.019. Epub 2016 Dec 5 [PubMed PMID: 27931865]
Huppertz A, Hermann KG, Diekhoff T, Wagner M, Hamm B, Schmidt WA. Systemic staging for urate crystal deposits with dual-energy CT and ultrasound in patients with suspected gout. Rheumatology international. 2014 Jun:34(6):763-71. doi: 10.1007/s00296-014-2979-1. Epub 2014 Mar 12 [PubMed PMID: 24619560]
Level 2 (mid-level) evidenceOgdie A, Taylor WJ, Weatherall M, Fransen J, Jansen TL, Neogi T, Schumacher HR, Dalbeth N. Imaging modalities for the classification of gout: systematic literature review and meta-analysis. Annals of the rheumatic diseases. 2015 Oct:74(10):1868-74. doi: 10.1136/annrheumdis-2014-205431. Epub 2014 Jun 10 [PubMed PMID: 24915980]
Level 1 (high-level) evidenceGarner HW, Wessell DE. Current status of ultrasound and dual-energy computed tomography in the evaluation of gout. Rheumatology international. 2018 Aug:38(8):1339-1344. doi: 10.1007/s00296-018-4033-1. Epub 2018 May 2 [PubMed PMID: 29721694]
Li S, Xu G, Liang J, Wan L, Cao H, Lin J. The Role of Advanced Imaging in Gout Management. Frontiers in immunology. 2021:12():811323. doi: 10.3389/fimmu.2021.811323. Epub 2022 Jan 14 [PubMed PMID: 35095904]
Lee YH, Song GG. Diagnostic accuracy of ultrasound in patients with gout: A meta-analysis. Seminars in arthritis and rheumatism. 2018 Apr:47(5):703-709. doi: 10.1016/j.semarthrit.2017.09.012. Epub 2017 Sep 28 [PubMed PMID: 29054295]
Level 1 (high-level) evidenceRichette P, Doherty M, Pascual E, Barskova V, Becce F, Castañeda-Sanabria J, Coyfish M, Guillo S, Jansen TL, Janssens H, Lioté F, Mallen C, Nuki G, Perez-Ruiz F, Pimentao J, Punzi L, Pywell T, So A, Tausche AK, Uhlig T, Zavada J, Zhang W, Tubach F, Bardin T. 2016 updated EULAR evidence-based recommendations for the management of gout. Annals of the rheumatic diseases. 2017 Jan:76(1):29-42. doi: 10.1136/annrheumdis-2016-209707. Epub 2016 Jul 25 [PubMed PMID: 27457514]
FitzGerald JD, Dalbeth N, Mikuls T, Brignardello-Petersen R, Guyatt G, Abeles AM, Gelber AC, Harrold LR, Khanna D, King C, Levy G, Libbey C, Mount D, Pillinger MH, Rosenthal A, Singh JA, Sims JE, Smith BJ, Wenger NS, Bae SS, Danve A, Khanna PP, Kim SC, Lenert A, Poon S, Qasim A, Sehra ST, Sharma TSK, Toprover M, Turgunbaev M, Zeng L, Zhang MA, Turner AS, Neogi T. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis care & research. 2020 Jun:72(6):744-760. doi: 10.1002/acr.24180. Epub 2020 May 11 [PubMed PMID: 32391934]
Schlesinger N, Detry MA, Holland BK, Baker DG, Beutler AM, Rull M, Hoffman BI, Schumacher HR Jr. Local ice therapy during bouts of acute gouty arthritis. The Journal of rheumatology. 2002 Feb:29(2):331-4 [PubMed PMID: 11838852]
Level 1 (high-level) evidenceKhanna D, Fitzgerald JD, Khanna PP, Bae S, Singh MK, Neogi T, Pillinger MH, Merill J, Lee S, Prakash S, Kaldas M, Gogia M, Perez-Ruiz F, Taylor W, Lioté F, Choi H, Singh JA, Dalbeth N, Kaplan S, Niyyar V, Jones D, Yarows SA, Roessler B, Kerr G, King C, Levy G, Furst DE, Edwards NL, Mandell B, Schumacher HR, Robbins M, Wenger N, Terkeltaub R, American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis care & research. 2012 Oct:64(10):1431-46. doi: 10.1002/acr.21772. Epub [PubMed PMID: 23024028]
Level 1 (high-level) evidenceZhang W, Doherty M, Bardin T, Pascual E, Barskova V, Conaghan P, Gerster J, Jacobs J, Leeb B, Lioté F, McCarthy G, Netter P, Nuki G, Perez-Ruiz F, Pignone A, Pimentão J, Punzi L, Roddy E, Uhlig T, Zimmermann-Gòrska I, EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Annals of the rheumatic diseases. 2006 Oct:65(10):1312-24 [PubMed PMID: 16707532]
Zhang W, Doherty M, Pascual E, Bardin T, Barskova V, Conaghan P, Gerster J, Jacobs J, Leeb B, Lioté F, McCarthy G, Netter P, Nuki G, Perez-Ruiz F, Pignone A, Pimentão J, Punzi L, Roddy E, Uhlig T, Zimmermann-Gòrska I, EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part I: Diagnosis. Report of a task force of the Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Annals of the rheumatic diseases. 2006 Oct:65(10):1301-11 [PubMed PMID: 16707533]
YU TF, GUTMAN AB. Study of the paradoxical effects of salicylate in low, intermediate and high dosage on the renal mechanisms for excretion of urate in man. The Journal of clinical investigation. 1959 Aug:38(8):1298-315 [PubMed PMID: 13673086]
YUE TF, DAYTON PG, GUTMAN AB. MUTUAL SUPPRESSION OF THE URICOSURIC EFFECTS OF SULFINPYRAZONE AND SALICYLATE: A STUDY IN INTERACTIONS BETWEEN DRUGS. The Journal of clinical investigation. 1963 Aug:42(8):1330-9 [PubMed PMID: 14060403]
Caspi D, Lubart E, Graff E, Habot B, Yaron M, Segal R. The effect of mini-dose aspirin on renal function and uric acid handling in elderly patients. Arthritis and rheumatism. 2000 Jan:43(1):103-8 [PubMed PMID: 10643705]
Segal R, Lubart E, Leibovitz A, Iaina A, Caspi D. Renal effects of low dose aspirin in elderly patients. The Israel Medical Association journal : IMAJ. 2006 Oct:8(10):679-82 [PubMed PMID: 17125112]
Zhang YK, Yang H, Zhang JY, Song LJ, Fan YC. Comparison of intramuscular compound betamethasone and oral diclofenac sodium in the treatment of acute attacks of gout. International journal of clinical practice. 2014 May:68(5):633-8. doi: 10.1111/ijcp.12359. Epub 2014 Jan 29 [PubMed PMID: 24472084]
Level 1 (high-level) evidenceAlloway JA, Moriarty MJ, Hoogland YT, Nashel DJ. Comparison of triamcinolone acetonide with indomethacin in the treatment of acute gouty arthritis. The Journal of rheumatology. 1993 Jan:20(1):111-3 [PubMed PMID: 8441139]
Level 1 (high-level) evidenceRainer TH, Cheng CH, Janssens HJ, Man CY, Tam LS, Choi YF, Yau WH, Lee KH, Graham CA. Oral Prednisolone in the Treatment of Acute Gout: A Pragmatic, Multicenter, Double-Blind, Randomized Trial. Annals of internal medicine. 2016 Apr 5:164(7):464-71. doi: 10.7326/M14-2070. Epub 2016 Feb 23 [PubMed PMID: 26903390]
Level 1 (high-level) evidenceYu J, Lu H, Zhou J, Xie Z, Wen C, Xu Z. Oral prednisolone versus non-steroidal anti-inflammatory drugs in the treatment of acute gout: a meta-analysis of randomized controlled trials. Inflammopharmacology. 2018 Jun:26(3):717-723. doi: 10.1007/s10787-018-0442-8. Epub 2018 Jan 22 [PubMed PMID: 29357007]
Level 1 (high-level) evidenceZeng L, Qasim A, Neogi T, Fitzgerald JD, Dalbeth N, Mikuls TR, Guyatt GH, Brignardello-Petersen R. Efficacy and Safety of Pharmacologic Interventions in Patients Experiencing a Gout Flare: A Systematic Review and Network Meta-Analysis. Arthritis care & research. 2021 May:73(5):755-764. doi: 10.1002/acr.24402. Epub [PubMed PMID: 32741131]
Level 1 (high-level) evidenceImazio M, Nidorf M. Colchicine and the heart. European heart journal. 2021 Jul 21:42(28):2745-2760. doi: 10.1093/eurheartj/ehab221. Epub [PubMed PMID: 33961006]
Terkeltaub RA, Furst DE, Bennett K, Kook KA, Crockett RS, Davis MW. High versus low dosing of oral colchicine for early acute gout flare: Twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis and rheumatism. 2010 Apr:62(4):1060-8. doi: 10.1002/art.27327. Epub [PubMed PMID: 20131255]
Level 1 (high-level) evidence. Colchicine and other drugs for gout. The Medical letter on drugs and therapeutics. 2009 Nov 30:51(1326):93-4 [PubMed PMID: 20224523]
Level 3 (low-level) evidenceTodd BA, Billups SJ, Delate T, Canty KE, Kauffman AB, Rawlings JE, Wagner TM. Assessment of the association between colchicine therapy and serious adverse events. Pharmacotherapy. 2012 Nov:32(11):974-80. doi: 10.1002/phar.1125. Epub 2012 Sep 27 [PubMed PMID: 23019065]
Level 2 (mid-level) evidenceTerkeltaub RA. Colchicine update: 2008. Seminars in arthritis and rheumatism. 2009 Jun:38(6):411-9. doi: 10.1016/j.semarthrit.2008.08.006. Epub 2008 Oct 29 [PubMed PMID: 18973929]
Kurup R, Galougahi KK, Figtree G, Misra A, Patel S. The Role of Colchicine in Atherosclerotic Cardiovascular Disease. Heart, lung & circulation. 2021 Jun:30(6):795-806. doi: 10.1016/j.hlc.2020.11.010. Epub 2021 Jan 16 [PubMed PMID: 33461916]
Deftereos SG, Beerkens FJ, Shah B, Giannopoulos G, Vrachatis DA, Giotaki SG, Siasos G, Nicolas J, Arnott C, Patel S, Parsons M, Tardif JC, Kovacic JC, Dangas GD. Colchicine in Cardiovascular Disease: In-Depth Review. Circulation. 2022 Jan 4:145(1):61-78. doi: 10.1161/CIRCULATIONAHA.121.056171. Epub 2021 Dec 29 [PubMed PMID: 34965168]
Solomon DH, Liu CC, Kuo IH, Zak A, Kim SC. Effects of colchicine on risk of cardiovascular events and mortality among patients with gout: a cohort study using electronic medical records linked with Medicare claims. Annals of the rheumatic diseases. 2016 Sep:75(9):1674-9. doi: 10.1136/annrheumdis-2015-207984. Epub 2015 Nov 18 [PubMed PMID: 26582823]
Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, Berry C, López-Sendón J, Ostadal P, Koenig W, Angoulvant D, Grégoire JC, Lavoie MA, Dubé MP, Rhainds D, Provencher M, Blondeau L, Orfanos A, L'Allier PL, Guertin MC, Roubille F. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. The New England journal of medicine. 2019 Dec 26:381(26):2497-2505. doi: 10.1056/NEJMoa1912388. Epub 2019 Nov 16 [PubMed PMID: 31733140]
Paulus HE, Schlosstein LH, Godfrey RG, Klinenberg JR, Bluestone R. Prophylactic colchicine therapy of intercritical gout. A placebo-controlled study of probenecid-treated patients. Arthritis and rheumatism. 1974 Sep-Oct:17(5):609-14 [PubMed PMID: 4606955]
Level 1 (high-level) evidenceBorstad GC, Bryant LR, Abel MP, Scroggie DA, Harris MD, Alloway JA. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. The Journal of rheumatology. 2004 Dec:31(12):2429-32 [PubMed PMID: 15570646]
Level 1 (high-level) evidenceLatourte A, Bardin T, Richette P. Prophylaxis for acute gout flares after initiation of urate-lowering therapy. Rheumatology (Oxford, England). 2014 Nov:53(11):1920-6. doi: 10.1093/rheumatology/keu157. Epub 2014 Apr 23 [PubMed PMID: 24758886]
Liew JW, Gardner GC. Use of Anakinra in Hospitalized Patients with Crystal-associated Arthritis. The Journal of rheumatology. 2019 Oct:46(10):1345-1349. doi: 10.3899/jrheum.181018. Epub 2019 Jan 15 [PubMed PMID: 30647192]
Saag KG, Khanna PP, Keenan RT, Ohlman S, Osterling Koskinen L, Sparve E, Åkerblad AC, Wikén M, So A, Pillinger MH, Terkeltaub R. A Randomized, Phase II Study Evaluating the Efficacy and Safety of Anakinra in the Treatment of Gout Flares. Arthritis & rheumatology (Hoboken, N.J.). 2021 Aug:73(8):1533-1542. doi: 10.1002/art.41699. Epub 2021 Jul 7 [PubMed PMID: 33605029]
Level 1 (high-level) evidenceSo A, De Meulemeester M, Pikhlak A, Yücel AE, Richard D, Murphy V, Arulmani U, Sallstig P, Schlesinger N. Canakinumab for the treatment of acute flares in difficult-to-treat gouty arthritis: Results of a multicenter, phase II, dose-ranging study. Arthritis and rheumatism. 2010 Oct:62(10):3064-76. doi: 10.1002/art.27600. Epub [PubMed PMID: 20533546]
Level 1 (high-level) evidenceSchlesinger N, Alten RE, Bardin T, Schumacher HR, Bloch M, Gimona A, Krammer G, Murphy V, Richard D, So AK. Canakinumab for acute gouty arthritis in patients with limited treatment options: results from two randomised, multicentre, active-controlled, double-blind trials and their initial extensions. Annals of the rheumatic diseases. 2012 Nov:71(11):1839-48. doi: 10.1136/annrheumdis-2011-200908. Epub 2012 May 14 [PubMed PMID: 22586173]
Level 1 (high-level) evidenceKiltz U, Smolen J, Bardin T, Cohen Solal A, Dalbeth N, Doherty M, Engel B, Flader C, Kay J, Matsuoka M, Perez-Ruiz F, da Rocha Castelar-Pinheiro G, Saag K, So A, Vazquez Mellado J, Weisman M, Westhoff TH, Yamanaka H, Braun J. Treat-to-target (T2T) recommendations for gout. Annals of the rheumatic diseases. 2017 Apr:76(4):632-638. doi: 10.1136/annrheumdis-2016-209467. Epub 2016 Sep 22 [PubMed PMID: 27658678]
Taylor TH, Mecchella JN, Larson RJ, Kerin KD, Mackenzie TA. Initiation of allopurinol at first medical contact for acute attacks of gout: a randomized clinical trial. The American journal of medicine. 2012 Nov:125(11):1126-1134.e7. doi: 10.1016/j.amjmed.2012.05.025. Epub [PubMed PMID: 23098865]
Level 1 (high-level) evidenceHill EM, Sky K, Sit M, Collamer A, Higgs J. Does starting allopurinol prolong acute treated gout? A randomized clinical trial. Journal of clinical rheumatology : practical reports on rheumatic & musculoskeletal diseases. 2015 Apr:21(3):120-5. doi: 10.1097/RHU.0000000000000235. Epub [PubMed PMID: 25807090]
Level 2 (mid-level) evidenceJia E, Zhang Y, Ma W, Li B, Geng H, Zhong L, Yao X, Xie J, Xiao Y, Jiang Y, Qiu X, Xiao M, Cui X, Wei J, Zhang J. Initiation of febuxostat for acute gout flare does not prolong the current episode: a randomized clinical trial. Rheumatology (Oxford, England). 2021 Sep 1:60(9):4199-4204. doi: 10.1093/rheumatology/keaa908. Epub [PubMed PMID: 33404656]
Level 1 (high-level) evidenceHung SI,Chung WH,Liou LB,Chu CC,Lin M,Huang HP,Lin YL,Lan JL,Yang LC,Hong HS,Chen MJ,Lai PC,Wu MS,Chu CY,Wang KH,Chen CH,Fann CS,Wu JY,Chen YT, HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proceedings of the National Academy of Sciences of the United States of America. 2005 Mar 15; [PubMed PMID: 15743917]
Stamp LK, Taylor WJ, Jones PB, Dockerty JL, Drake J, Frampton C, Dalbeth N. Starting dose is a risk factor for allopurinol hypersensitivity syndrome: a proposed safe starting dose of allopurinol. Arthritis and rheumatism. 2012 Aug:64(8):2529-36. doi: 10.1002/art.34488. Epub [PubMed PMID: 22488501]
Level 2 (mid-level) evidenceStamp LK, O'Donnell JL, Zhang M, James J, Frampton C, Barclay ML, Chapman PT. Using allopurinol above the dose based on creatinine clearance is effective and safe in patients with chronic gout, including those with renal impairment. Arthritis and rheumatism. 2011 Feb:63(2):412-21. doi: 10.1002/art.30119. Epub [PubMed PMID: 21279998]
Sarawate CA, Patel PA, Schumacher HR, Yang W, Brewer KK, Bakst AW. Serum urate levels and gout flares: analysis from managed care data. Journal of clinical rheumatology : practical reports on rheumatic & musculoskeletal diseases. 2006 Apr:12(2):61-5 [PubMed PMID: 16601538]
Level 2 (mid-level) evidenceRagab AH, Gilkerson E, Myers M. The effect of 6-mercaptopurine and allopurinol on granulopoiesis. Cancer research. 1974 Sep:34(9):2246-9 [PubMed PMID: 4843532]
Level 3 (low-level) evidenceBecker MA, Schumacher HR Jr, Wortmann RL, MacDonald PA, Eustace D, Palo WA, Streit J, Joseph-Ridge N. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. The New England journal of medicine. 2005 Dec 8:353(23):2450-61 [PubMed PMID: 16339094]
Level 1 (high-level) evidenceBecker MA, Schumacher HR, MacDonald PA, Lloyd E, Lademacher C. Clinical efficacy and safety of successful longterm urate lowering with febuxostat or allopurinol in subjects with gout. The Journal of rheumatology. 2009 Jun:36(6):1273-82. doi: 10.3899/jrheum.080814. Epub 2009 Mar 13 [PubMed PMID: 19286847]
White WB, Saag KG, Becker MA, Borer JS, Gorelick PB, Whelton A, Hunt B, Castillo M, Gunawardhana L, CARES Investigators. Cardiovascular Safety of Febuxostat or Allopurinol in Patients with Gout. The New England journal of medicine. 2018 Mar 29:378(13):1200-1210. doi: 10.1056/NEJMoa1710895. Epub 2018 Mar 12 [PubMed PMID: 29527974]
Su CY, Shen LJ, Hsieh SC, Lin LY, Lin FJ. Comparing Cardiovascular Safety of Febuxostat and Allopurinol in the Real World: A Population-Based Cohort Study. Mayo Clinic proceedings. 2019 Jul:94(7):1147-1157. doi: 10.1016/j.mayocp.2019.03.001. Epub [PubMed PMID: 31272565]
Weisshaar S, Litschauer B, Reichardt B, Gruber F, Leitner S, Sibinovic S, Kossmeier M, Wolzt M. Cardiovascular risk and mortality in patients with hyperuricemia treated with febuxostat or allopurinol: a retrospective nation-wide cohort study in Austria 2014-2017. Rheumatology international. 2022 Sep:42(9):1597-1603. doi: 10.1007/s00296-022-05139-8. Epub 2022 May 19 [PubMed PMID: 35589988]
Level 2 (mid-level) evidenceMackenzie IS, Ford I, Nuki G, Hallas J, Hawkey CJ, Webster J, Ralston SH, Walters M, Robertson M, De Caterina R, Findlay E, Perez-Ruiz F, McMurray JJV, MacDonald TM, FAST Study Group. Long-term cardiovascular safety of febuxostat compared with allopurinol in patients with gout (FAST): a multicentre, prospective, randomised, open-label, non-inferiority trial. Lancet (London, England). 2020 Nov 28:396(10264):1745-1757. doi: 10.1016/S0140-6736(20)32234-0. Epub 2020 Nov 9 [PubMed PMID: 33181081]
Level 1 (high-level) evidenceKang EH, Choi HK, Shin A, Lee YJ, Lee EB, Song YW, Kim SC. Comparative cardiovascular risk of allopurinol versus febuxostat in patients with gout: a nation-wide cohort study. Rheumatology (Oxford, England). 2019 Dec 1:58(12):2122-2129. doi: 10.1093/rheumatology/kez189. Epub [PubMed PMID: 31098635]
Level 2 (mid-level) evidenceJeong H, Choi E, Suh A, Yoo M, Kim B. `Risk of cardiovascular disease associated with febuxostat versus allopurinol use in patients with gout: a retrospective cohort study in Korea. Rheumatology international. 2023 Feb:43(2):265-281. doi: 10.1007/s00296-022-05222-0. Epub 2022 Nov 8 [PubMed PMID: 36346443]
Level 2 (mid-level) evidenceGao L, Wang B, Pan Y, Lu Y, Cheng R. Cardiovascular safety of febuxostat compared to allopurinol for the treatment of gout: A systematic and meta-analysis. Clinical cardiology. 2021 Jul:44(7):907-916. doi: 10.1002/clc.23643. Epub 2021 May 20 [PubMed PMID: 34013998]
Level 1 (high-level) evidenceGhang BZ, Lee JS, Choi J, Kim J, Yoo B. Increased risk of cardiovascular events and death in the initial phase after discontinuation of febuxostat or allopurinol: another story of the CARES trial. RMD open. 2022 Jun:8(2):. doi: 10.1136/rmdopen-2021-001944. Epub [PubMed PMID: 35732345]
O'Dell JR, Brophy MT, Pillinger MH, Neogi T, Palevsky PM, Wu H, Davis-Karim A, Newcomb JA, Ferguson R, Pittman D, Cannon GW, Taylor T, Terkeltaub R, Cannella AC, England BR, Helget LN, Mikuls TR. Comparative Effectiveness of Allopurinol and Febuxostat in Gout Management. NEJM evidence. 2022 Mar:1(3):. doi: 10.1056/evidoa2100028. Epub 2022 Feb 3 [PubMed PMID: 35434725]
Level 2 (mid-level) evidenceGupta MK, Singh JA. Cardiovascular Disease in Gout and the Protective Effect of Treatments Including Urate-Lowering Therapy. Drugs. 2019 Apr:79(5):531-541. doi: 10.1007/s40265-019-01081-5. Epub [PubMed PMID: 30868398]
Dubreuil M, Zhu Y, Zhang Y, Seeger JD, Lu N, Rho YH, Choi HK. Allopurinol initiation and all-cause mortality in the general population. Annals of the rheumatic diseases. 2015 Jul:74(7):1368-72. doi: 10.1136/annrheumdis-2014-205269. Epub 2014 Mar 24 [PubMed PMID: 24665118]
Level 2 (mid-level) evidenceLuk AJ,Levin GP,Moore EE,Zhou XH,Kestenbaum BR,Choi HK, Allopurinol and mortality in hyperuricaemic patients. Rheumatology (Oxford, England). 2009 Jul [PubMed PMID: 19447769]
Level 2 (mid-level) evidenceChen JH, Lan JL, Cheng CF, Liang WM, Lin HY, Tsay GJ, Yeh WT, Pan WH. Effect of Urate-lowering Therapy on the Risk of Cardiovascular Disease and All-cause Mortality in Patients with Gout: A Case-matched Cohort Study. The Journal of rheumatology. 2015 Sep:42(9):1694-701. doi: 10.3899/jrheum.141542. Epub 2015 Jun 15 [PubMed PMID: 26077411]
Level 3 (low-level) evidenceSingh JA, Yu S. Allopurinol and the risk of atrial fibrillation in the elderly: a study using Medicare data. Annals of the rheumatic diseases. 2017 Jan:76(1):72-78. doi: 10.1136/annrheumdis-2015-209008. Epub 2016 May 10 [PubMed PMID: 27165177]
Vargas-Santos AB, Peloquin CE, Zhang Y, Neogi T. Association of Chronic Kidney Disease With Allopurinol Use in Gout Treatment. JAMA internal medicine. 2018 Nov 1:178(11):1526-1533. doi: 10.1001/jamainternmed.2018.4463. Epub [PubMed PMID: 30304329]
Singh JA, Yu S. Are allopurinol dose and duration of use nephroprotective in the elderly? A Medicare claims study of allopurinol use and incident renal failure. Annals of the rheumatic diseases. 2017 Jan:76(1):133-139. doi: 10.1136/annrheumdis-2015-209046. Epub 2016 Jun 13 [PubMed PMID: 27296322]
Levy GD, Rashid N, Niu F, Cheetham TC. Effect of urate-lowering therapies on renal disease progression in patients with hyperuricemia. The Journal of rheumatology. 2014 May:41(5):955-62. doi: 10.3899/jrheum.131159. Epub 2014 Apr 1 [PubMed PMID: 24692523]
Level 2 (mid-level) evidenceKim Y, Shin S, Kim K, Choi S, Lee K. Effect of Urate Lowering Therapy on Renal Disease Progression in Hyperuricemic Patients with Chronic Kidney Disease. The Journal of rheumatology. 2015 Nov:42(11):2143-8. doi: 10.3899/jrheum.150067. Epub 2015 Oct 1 [PubMed PMID: 26428209]
Singh JA, Cleveland JD. Comparative effectiveness of allopurinol versus febuxostat for preventing incident renal disease in older adults: an analysis of Medicare claims data. Annals of the rheumatic diseases. 2017 Oct:76(10):1669-1678. doi: 10.1136/annrheumdis-2017-211210. Epub 2017 Jun 5 [PubMed PMID: 28584186]
Level 2 (mid-level) evidenceWeinberger A, Schindel B, Liberman UA, Pinkhas J, Sperling O. Calciuric effect of probenecid in gouty patients. Israel journal of medical sciences. 1983 Apr:19(4):377-9 [PubMed PMID: 6853140]
Schlesinger N, Yasothan U, Kirkpatrick P. Pegloticase. Nature reviews. Drug discovery. 2011 Jan:10(1):17-8. doi: 10.1038/nrd3349. Epub [PubMed PMID: 21193861]
Level 3 (low-level) evidenceSundy JS, Baraf HS, Yood RA, Edwards NL, Gutierrez-Urena SR, Treadwell EL, Vázquez-Mellado J, White WB, Lipsky PE, Horowitz Z, Huang W, Maroli AN, Waltrip RW 2nd, Hamburger SA, Becker MA. Efficacy and tolerability of pegloticase for the treatment of chronic gout in patients refractory to conventional treatment: two randomized controlled trials. JAMA. 2011 Aug 17:306(7):711-20. doi: 10.1001/jama.2011.1169. Epub [PubMed PMID: 21846852]
Level 1 (high-level) evidenceBecker MA, Baraf HS, Yood RA, Dillon A, Vázquez-Mellado J, Ottery FD, Khanna D, Sundy JS. Long-term safety of pegloticase in chronic gout refractory to conventional treatment. Annals of the rheumatic diseases. 2013 Sep 1:72(9):1469-74. doi: 10.1136/annrheumdis-2012-201795. Epub 2012 Nov 10 [PubMed PMID: 23144450]
Level 1 (high-level) evidenceKeenan RT, Yeo AE, Lipsky PE. Pegloticase causes prolonged improvement in multiple disease parameters in patients with chronic refractory gout who maintain low serum urate levels. Clinical and experimental rheumatology. 2022 May:40(5):1006-1010. doi: 10.55563/clinexprheumatol/3m095f. Epub 2022 Feb 25 [PubMed PMID: 35238750]
Level 3 (low-level) evidenceLeung N, Yip K, Pillinger MH, Toprover M. Lowering and Raising Serum Urate Levels: Off-Label Effects of Commonly Used Medications. Mayo Clinic proceedings. 2022 Jul:97(7):1345-1362. doi: 10.1016/j.mayocp.2022.02.027. Epub [PubMed PMID: 35787862]
Zhao Y, Xu L, Tian D, Xia P, Zheng H, Wang L, Chen L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes, obesity & metabolism. 2018 Feb:20(2):458-462. doi: 10.1111/dom.13101. Epub 2017 Sep 27 [PubMed PMID: 28846182]
Level 1 (high-level) evidenceDoehner W, Anker SD, Butler J, Zannad F, Filippatos G, Ferreira JP, Salsali A, Kaempfer C, Brueckmann M, Pocock SJ, Januzzi JL, Packer M. Uric acid and sodium-glucose cotransporter-2 inhibition with empagliflozin in heart failure with reduced ejection fraction: the EMPEROR-reduced trial. European heart journal. 2022 Sep 21:43(36):3435-3446. doi: 10.1093/eurheartj/ehac320. Epub [PubMed PMID: 35788657]
Fralick M, Chen SK, Patorno E, Kim SC. Assessing the Risk for Gout With Sodium-Glucose Cotransporter-2 Inhibitors in Patients With Type 2 Diabetes: A Population-Based Cohort Study. Annals of internal medicine. 2020 Feb 4:172(3):186-194. doi: 10.7326/M19-2610. Epub 2020 Jan 14 [PubMed PMID: 31931526]
Berman EL. Clues in the eye: ocular signs of metabolic and nutritional disorders. Geriatrics. 1995 Jul:50(7):34-6, 43-4 [PubMed PMID: 7601360]
Choi HK, Atkinson K, Karlson EW, Curhan G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the health professionals follow-up study. Archives of internal medicine. 2005 Apr 11:165(7):742-8 [PubMed PMID: 15824292]
Roubenoff R, Klag MJ, Mead LA, Liang KY, Seidler AJ, Hochberg MC. Incidence and risk factors for gout in white men. JAMA. 1991 Dec 4:266(21):3004-7 [PubMed PMID: 1820473]
Level 2 (mid-level) evidenceMartinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006 Mar 9:440(7081):237-41 [PubMed PMID: 16407889]
Level 3 (low-level) evidenceSingh JA. Gout and comorbidity: a nominal group study of people with gout. Arthritis research & therapy. 2017 Sep 15:19(1):204. doi: 10.1186/s13075-017-1416-8. Epub 2017 Sep 15 [PubMed PMID: 28915838]