
Introduction
Acromegaly and gigantism are disorders of growth hormone hypersecretion. The most common cause is a growth hormone (GH) secreting adenoma in the pituitary gland. Gigantism occurs when growth hormone hypersecretion occurs before the fusion of the long bone epiphysis and is characterized by tall stature. Acromegaly occurs when GH hypersecretion occurs after the fusion of the epiphysis leading to large extremities and characteristic facies. An elevated insulin-like growth factor-1 (IGF-1) level establishes the diagnosis. The first line of treatment is surgical excision of the tumor; however, this rarely results in a cure, and further medical treatment with somatostatin analogs or radiation is usually necessary.[1][2]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
About 95% of the cases of acromegaly and gigantism are secondary to a GH-secreting adenoma in the pituitary gland. Growth hormone-releasing hormone (GHRH) secretion from a hypothalamic adenoma or ectopic GHRH secretion from lung or pancreas neuroendocrine tumors can also cause acromegaly. Rarely, ectopic growth hormone secretion secondary to abdominal and hemopoietic malignancies can cause acromegaly. Genetic syndromes associated with GH hypersecretion are multiple endocrine neoplasia-1 (MEN-1), neurofibromatosis, Carney complex, and McCune-Albright syndrome. Familial idiopathic pituitary adenomas (FIPA), due to aryl hydrocarbon protein-interacting (AIP) mutations, can be associated with familial acromegaly. About 25% of the cases with familial acromegaly usually present as teenagers with gigantism.[3]
Epidemiology
The prevalence of acromegaly is 78 cases per million population, and the incidence is ten new cases per year per million population.[4] There is no gender preponderance with equal incidence in males and females. The average age of presentation is 44 years, with younger patients tending to have more aggressive disease. About 33% of the cases of acromegaly have co-existent hyperprolactinemia.[5]
Pathophysiology
GH is a 191 amino acid-long protein with two disulfide bonds. It is secreted by the somatotroph cells in the anterior pituitary. It circulates for the most part as a 22kD protein and the remaining as a 20kD protein. It is secreted in a pulsatile manner, 4 to 11 pulses in a day. Due to this pulsatile nature of secretion, the measurement of random GH levels is not useful. Growth hormone-releasing hormone (GHRH) stimulates the release of GH from the pituitary. GHRH-containing neurons are mainly seen in the arcuate nucleus and ventromedial nucleus. Somatostatin, which is also secreted from the hypothalamus, exerts an inhibitory action on the secretion of GH. GHRH and somatostatin also regulate each other's secretion in a paracrine manner. GH stimulates the synthesis of IGF-1 from the liver. IGF-1 is a 70 amino acid protein that is similar to insulin. Besides, post-receptor signaling mechanisms involving tyrosine kinase and insulin receptor substrate-1 (IRS-1) are also similar for IGF-1 and insulin. IGF-1 circulates bound to IGF-1 binding proteins. IGF-1 exerts a negative feedback mechanism through GHRH and somatostatin.
Several other hormones can also modulate the secretion of GH. The thyroid hormone facilitates the expression of the growth hormone gene. Hypothyroidism has been known to be associated with low GH and IGF-1, which are reversible with thyroid hormone replacement therapy. Gonadal hormones can also upregulate the secretion of GH, as evident during the onset of puberty. Hypoglycemia decreases somatostatin secretion from the hypothalamus and increases the release of GH. Mutations in the somatotrophs are a prerequisite for the abnormal response to GHRH. Point mutations in Arg 201 and Gly227 have been reported, which are the sites for adenosine diphosphate (ADP) ribosylation and the binding domain of guanine triphosphate (GTP), respectively, and they result in adenyl cyclase activation similar to GHRH. The tumorigenesis, which leads to a monoclonal cell expansion, is multifactorial, and the presence of a mutation alone is not enough.[6]
Histopathology
Densely Granulated GH Cell Adenomas
These constitute about 30% of GH-secreting pituitary adenomas, are characterized by cells similar to somatotrophs, and contain numerous large secretory granules. These are slow-growing tumors that present in middle age.
Sparsely Granulated GH Cell Adenomas
These constitute about 30% of GH-secreting pituitary adenomas and are characterized by pleomorphic cells with few secretory granules. These are rapidly growing tumors that present with severe disease at a young age.
Mixed GH Cell and Prolactin Cell Adenoma
These are about 20% of GH-secreting pituitary adenomas, characterized by densely granulated somatotrophs and sparsely granulated lactotrophs. They secrete GH and prolactin.
Acidophilic Stem Cell Adenomas
These are rare tumors and are characterized by giant mitochondria. Clinically these are aggressive tumors that invade the surrounding structures. They also secrete GH and prolactin.
Mammosomatotroph Cell Adenoma
These comprise about 10% of GH-secreting pituitary adenomas and are characterized by cells that secrete both GH and prolactin. They commonly occur in children and present with gigantism and hyperprolactinemia.
Plurihormonal Cell Adenomas
As the name suggests, these tumors may secrete multiple hormones GH, prolactin, follicle-stimulating hormone (FSH), Thyrotropin stimulating hormone (TSH), or Adrenocorticotrophic hormone (ACTH); the secondary hormonal products may be silent.[7][8]
History and Physical
Acromegaly is a very insidious disease where changes occur over many years. Consequently, studies have reported that the diagnosis is often delayed for about nine years on average. Patients may seek care for dental problems and cardiac or rheumatological issues before being referred to endocrinology.
GH-secreting pituitary adenomas are often big (> 1 cm) and can have compressive symptoms in the form of visual field deficits, ophthalmoplegia, and headaches, which are out of proportion to the size of the tumor. Co-existent hyperprolactinemia can result in galactorrhea and symptoms of hypogonadism (irregular periods in females and decreased libido in males).
Acromegaly Characteristics
Increased size of extremities: Enlargement of the hands and feet is noted secondary to both bony expansion and soft tissue swelling. Patients often appreciate an increase in ring and shoe size. The extremities have a "dough" like consistency due to the soft tissue swelling. There is a decrease in the shoe size and ring size with the treatment of acromegaly due to the resolution of soft tissue swelling; the bony changes, however, are permanent.
Hyperhidrosis and skin tags are present in about 98% of cases of acromegaly; these features are sensitive signs for detecting acromegaly. Skin tags are due to the epithelial cell hyperproliferation induced by GH.
Acromegalic facies: Prominent supraorbital ridge, broad nose, acne, large lips, overbite, prognathism, tongue enlargement, and coarsening of facial features form the characteristic acromegalic facies. These changes, however, are very subtle and often go unrecognized by family members. A comparison of present and old photographs may show gradual changes and is an essential part of the assessment of acromegaly.
Musculoskeletal: Generalized weakness and lethargy are common symptoms. Elongation of the jaw can lead to teeth malocclusion, temporomandibular joint pain, and a characteristic interdental separation. Carpal tunnel syndrome is seen in about 60% of patients and is secondary to median nerve swelling rather than extrinsic compression. Early-onset osteoarthritis may be seen due to incongruent articular surfaces in the hips, knees, and spine. Kyphoscoliosis has also been reported in association with acromegaly.
Deep voice and obstructive sleep apnea can occur secondary to soft tissue swelling of the upper airway and large tongue.[9][10][11]
Gigantism
Gigantism is very rare and should be suspected when the patient's height is 3 standard deviations above normal mean height or 2 standard deviations above the adjusted mean parental height. Since gigantism is associated with various syndromes like neurofibromatosis, Carney complex, and McCune Albright syndrome, evaluation for neurofibromas with cafe au lait spots, optic gliomas, and skin lentigines should be done.[12]
Evaluation
Biochemical diagnosis: Measurement of IGF-1 level is the initial test for diagnosing acromegaly as it is a stable molecule with a half-life of 15 hours. It should be measured in cases with clinical suspicion of acromegaly and pituitary masses – normal IGF-1 level rules out acromegaly. False-positive IGF-1 levels can be seen in pregnancy and adolescence, and false-negative levels may be seen with estrogen therapy. Furthermore, hepatic failure, renal failure, hypothyroidism, malnutrition, sepsis, and poorly controlled diabetes mellitus can also influence IGF-1 levels. All cases with elevated IGF-1 levels need to have an oral glucose tolerance test (OGTT) with GH measurement to confirm the diagnosis of acromegaly or gigantism. A GH level of 1 mcg/lt or less, 2 hours after a 75 gms of oral glucose tolerance test, rules out acromegaly. Plasma glucose needs to be measured before and after the administration of glucose to make sure hyperglycemia has been achieved.[13]
Imaging: Pituitary Magnetic resonance imaging (MRI) is the preferred imaging modality for diagnosing acromegaly. The size, extent of the tumor, optic chiasmal compression, and cavernous sinus invasion can all be assessed on the MRI scan. Visual field testing is to be done in all cases where the tumor is in contact with the optic chiasm on the MRI scan.
Other tests: Prolactin levels need to be assessed especially in the presence of galactorrhea or symptoms of hypogonadism. Anterior pituitary hormonal assessment needs to be done based on the clinical picture. Rarely, in the presence of normal pituitary and biochemically confirmed acromegaly, GHRH levels and imaging of the chest and abdomen need to be done to evaluate for ectopic GH or GHRH secretion.[1]
Treatment / Management
The aims of treatment in acromegaly are:
- Removal of the pituitary tumor to relieve mass effects
- Normalization of IGF-1 levels
- Relief of acromegalic symptoms
- Monitoring of acromegaly and associated comorbidities[6]
Three primary modalities are available for treating acromegaly, each with its advantages and disadvantages: surgery, medical therapy, and radiation. The decision to use these modalities is made on a case-by-case basis.
Surgery
Surgical excision of the tumor is the preferred initial treatment unless the patient is deemed unfit for surgery. Surgery is also the preferred modality in case of recurrence as long as the tumor remains accessible. The transsphenoidal approach involves accessing the tumor by getting to the sphenoid sinus either through a nasal or sublabial approach and removing the sellar floor. Even tumors with suprasellar extension can be removed with this approach. Endoscopic transsphenoidal resection has better tumor clearance, decreased morbidity, and complications compared to the microscopic approach. Post-surgical complications include diabetes insipidus and anterior pituitary hormone deficiencies.[14][15](B3)
IGF-1 and GH levels need to be measured 12 weeks after surgery. The goal is a normalization of IGF-1 and an undetectable GH level.
Medical Treatment
Various medical therapies are available for the management of acromegaly. They are used in the treatment of persistent disease after surgery.
- Dopamine agonists: Cabergoline is a D2 receptor agonist that can act on the D2 receptors in somatotrophs and decrease GH secretion. However, their efficacy is limited, with normalization of IGF-1 seen only in 34% of patients. Side effects of cabergoline include dizziness, nausea, vomiting, and postural hypotension.[16]
- Somatostatin receptor ligands (SRL): These mimic the action of somatostatin and inhibit the secretion of GH. Octreotide was the first SRL to be introduced, and it is 0preferentially bound to human somatostatin receptor type 2. The dose was 100 to 200 mg by subcutaneous injection three times a day. Long-acting SRL has become available in the form of octreotide LAR and lanreotide. Both have similar efficacy in suppressing GH levels and normalizing IGF-1 levels. Octreotide LAR is given at a dose of 10 to 30 mg subcutaneously every four weeks, while lanreotide is given at a dose of 120 mg subcutaneously every four weeks. Recently, an oral formulation of the somatostatin approved showed equivalent efficacy to the injectable forms.[17][18] Side effects of SRL include abdominal pain, nausea, flatulence, diarrhea, and hyperglycemia. The primary role of SRL is as an adjunct to radiation after surgery; however, in certain instances, it may be used to decrease the tumor size or improve cardiovascular function preoperatively.[19][20] A second-generation somatostatin analog includes Pasireotide.[21][22][23]
- GH receptor antagonists: GH binds to its receptor on two different sites and induces dimerization, activating the post-receptor signaling mechanisms. The novel GH receptor antagonist pegvisomant binds to the first binding site, preventing dimerization and post-receptor signaling. Van der Lely et al. followed 152 patients treated with pegvisomant for 18 months and found normalization of IGF-1 in 90% of patients. Pegvisomant is administered at a dose of 10 to 30 mg subcutaneously every day. Due to the high expense, its role is limited to patients who do not respond to SRL or have diabetes and worsening hyperglycemia with SRL.[24][25][26] (A1)
Radiation Therapy
Radiation is often used as an adjunct treatment for the persistent disease after surgery; rarely, it may be used as a first-line treatment in patients unfit for surgery. Two commonly used radiation therapy modalities are external irradiation and stereotactic single high-dose irradiation.
- External irradiation: Using linear accelerators, the radiation is focused on the pituitary fossa. CT or MRI are used for dosimetry to minimize radiation exposure to the surrounding tissues. The total radiation dose (around 4500 cGy) is divided into fractions (generally 25 fractions of 180 cGy) and given over six weeks; this fractionation helps minimize injury to surrounding tissues. About 60% of the patients achieve normalization of IGF-1 with radiation in 10 years. Hypopituitarism is a known side effect of radiation and can occur years after cessation of radiation; therefore, annual clinical and laboratory assessments for pituitary hormonal deficiencies should be done. With external irradiation, there is a resolution of even radiologically invisible disease.
- Stereotactic single high dose irradiation: Using a gamma knife or stereotactic multiple arc radiotherapy, a high dose of radiation is delivered to a previously mapped area. Care should be taken to limit radiation exposure (< 8 cGy) to the optic chiasm. Since the radiation is delivered to a mapped area, any radiologically invisible disease will not be resolved. The rate of development of hypopituitarism is the same as that of external irradiation.[27][28] (B2)
Differential Diagnosis
Acromegaloidism: This is a condition where the patients have acromegaloid facial features or tall stature; however, laboratory assessments of GH and IGF-1 are normal. Imaging of the pituitary in these cases is unremarkable.[29]
Soto's syndrome: This is a congenital overgrowth syndrome characterized by tall stature, acromegaloid facies, intellectual disabilities, macrocephaly, and advanced bone age. Other clinical features include neonatal hypotonia, congenital heart defects, strabismus, scoliosis, and a predisposition to cancer. Soto syndrome is due to the haploinsufficiency of the NSD1 gene on chromosome 5. Laboratory assessment of IGF-1 and GH levels is normal. Genetic studies are needed to differentiate it from acromegaly.[30]
Prognosis
The prognosis depends on the stage at which the diagnosis is made, as well as the response of hormone levels to treatment, either surgical or non-surgical.
Complications
Cardiovascular Complications
- Hypertension is seen in about 40% of patients with acromegaly and is usually mild. Anti-hypertensive treatment is similar to non-acromegalic patients. Good control of blood pressure is very important irrespective of the modality used for acromegalic treatment.
- Cardiomyopathy is seen in most patients with acromegaly. An echocardiogram and electrocardiogram (ECG) should be done at baseline and repeated yearly. Treatment of acromegaly improves cardiomyopathy; however, this depends on the patient's age, disease duration, and hypertension.[31] In addition to the echocardiogram and ECG, patients with gigantism will need Doppler of the peripheral arteries and veins.
Obstructive Sleep Apnea (OSA)
The prevalence of sleep apnea is 70% of all patients with acromegaly. Prognathism, enlarged tongue, and soft tissue accumulation in the upper airways predispose to OSA. Clinical assessment (Epworth score) and, if needed, polysomnography should be done at baseline and repeated every year. Surgical correction of prognathism may help, and referral to the maxillofacial surgeon should be considered.
Arthropathy
Around 75% of patients with acromegaly are affected by arthropathy. Both small and large joints are affected. Bony expansion and soft tissue swelling can lead to nerve entrapment. Early diagnosis and aggressive treatment of acromegaly are essential to prevent arthropathy, as these changes are irreversible.
Colon Polyps
Colon length is increased in acromegaly, and so are the mucosal folds. There is an increased prevalence of colonic polyps; however, the risk of colon cancer may or may not be increased. Patients should get a colonoscopy at baseline and every five years.[32]
The incidence of both benign and malignant tumors in patients with acromegaly has increased lately, like kidney and ureteral cancers, but not the mortality from cancers.[33]
Hypopituitarism can occur as a result of surgery or radiation. An annual assessment of pituitary hormones is recommended, and replacement hormones are needed.[34]
Vertebral fractures have been reported more frequently in patients with acromegaly. In a recent study, there seems to be a correlation between a hypogonadal state and bone loss in this population.[35][36][37]
Deterrence and Patient Education
Patients should be educated on the importance of early diagnosis and adhering to the treatment. They should undergo regular endocrine evaluations and check their blood pressure and their blood sugar regularly.
Pearls and Other Issues
- Acromegaly and gigantism are due to the oversecretion of growth hormone. The most common cause is a GH-secreting pituitary adenoma. Rarely, ectopic GH secretion or excess secretion of GHRH may be the cause.
- Gigantism is characterized by tall stature and should be suspected in children three standard deviations above the mean. Acromegaly is characterized by large hands and feet, coarse facial features, a broad nose, acne, hyperhidrosis, underbite, and teeth separation.[38]
- Laboratory assessment includes measurement of IGF-1 level; normal value rules out acromegaly.
- If IGF-1 is high, GH levels are assessed before and after OGTT to confirm the diagnosis.
- Surgery is the preferred initial modality of treatment. Residual or persistent findings can be treated medically or with radiotherapy.
- Medical treatment involves using somatostatin analogs, GH receptor antagonists, or dopamine receptor agonists.
- Radiotherapy involves either the use of external irradiation or stereotactic single high-dose irradiation, depending on the location and extent of residual disease.
- Mortality is increased in untreated acromegaly, with cardiovascular disease being the major contributor, but the quality of life of these patients has generally improved in recent years.[39]
- Besides control of GH, the management of acromegaly involves the treatment of acromegalic complications, which include hypertension, cardiomyopathy, OSA, colonic polyps, hypopituitarism, and arthropathy.
Enhancing Healthcare Team Outcomes
The management of acromegaly and gigantism should be done with an interprofessional team that includes an endocrinologist, neurosurgeon, internist, cardiologist, rheumatologist, pulmonologist, oncologist, nursing staff, and pharmacists. Clinicians, including specialists and mid-level practitioners, will guide the overall direction of care, but interprofessional care coordination and open communication are crucial to successful case management. Nurses will often help coordinate the activities of the clinicians, as well as assist in patient evaluation during surgical procedures and provide patient counsel. Pharmacists are vital resources for the pharmacological management of the disease, verifying agent selection and dosing, performing medication reconciliation, and counseling patients on the proper use of their medication and wheat adverse events might present. Any team member who notes a change in patient status should communicate their findings to the rest of the team and document their observations in the patient's medical record. This interprofessional approach to care will yield the most effective therapy delivery and improve patient outcomes. [Level 5]
Untreated acromegaly has increased mortality compared to the general population. The major contributors to mortality are cardiovascular disease (60%), respiratory disease (25%), and malignancies (15%). Mortality rates are similar to the general population in patients with controlled GH secretion. Besides the level of GH, the presence of cardiac disease is an important prognostic indicator. The presence of cardiac disease at diagnosis has a 100% mortality rate at 15 years. The mortality rate of patients with acromegaly and diabetes at 20 years is 80%.[40][41]
Media
(Click Image to Enlarge)
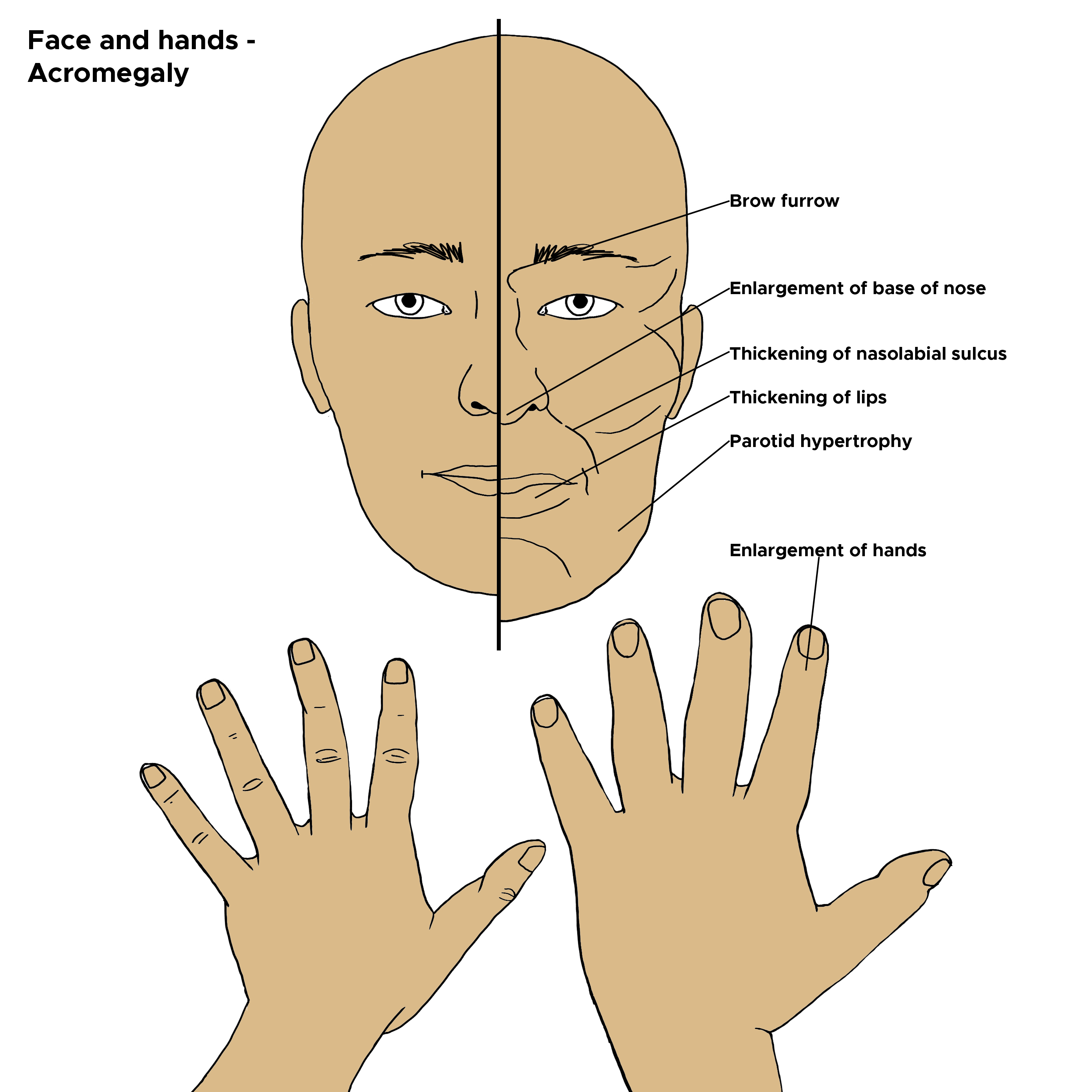
Acromegaly, gigantism. Illustrated image of physical attributes of acromegaly of the face and hands.
Contributed by C Rowe
References
Katznelson L, Laws ER Jr, Melmed S, Molitch ME, Murad MH, Utz A, Wass JA, Endocrine Society. Acromegaly: an endocrine society clinical practice guideline. The Journal of clinical endocrinology and metabolism. 2014 Nov:99(11):3933-51. doi: 10.1210/jc.2014-2700. Epub 2014 Oct 30 [PubMed PMID: 25356808]
Level 1 (high-level) evidenceFleseriu M,Biller BMK,Freda PU,Gadelha MR,Giustina A,Katznelson L,Molitch ME,Samson SL,Strasburger CJ,van der Lely AJ,Melmed S, A Pituitary Society update to acromegaly management guidelines. Pituitary. 2021 Feb [PubMed PMID: 33079318]
Adelman DT,Liebert KJ,Nachtigall LB,Lamerson M,Bakker B, Acromegaly: the disease, its impact on patients, and managing the burden of long-term treatment. International journal of general medicine. 2013 [PubMed PMID: 23359786]
Burton T,Le Nestour E,Neary M,Ludlam WH, Incidence and prevalence of acromegaly in a large US health plan database. Pituitary. 2016 Jun; [PubMed PMID: 26792654]
Holdaway IM, Rajasoorya C. Epidemiology of acromegaly. Pituitary. 1999 Jun:2(1):29-41 [PubMed PMID: 11081170]
Carroll PV,Jenkins PJ,Feingold KR,Anawalt B,Boyce A,Chrousos G,de Herder WW,Dhatariya K,Dungan K,Hershman JM,Hofland J,Kalra S,Kaltsas G,Koch C,Kopp P,Korbonits M,Kovacs CS,Kuohung W,Laferrère B,Levy M,McGee EA,McLachlan R,Morley JE,New M,Purnell J,Sahay R,Singer F,Sperling MA,Stratakis CA,Trence DL,Wilson DP, Acromegaly Endotext. 2000 [PubMed PMID: 25905322]
Melmed S, Medical progress: Acromegaly. The New England journal of medicine. 2006 Dec 14; [PubMed PMID: 17167139]
Melmed S,Braunstein GD,Horvath E,Ezrin C,Kovacs K, Pathophysiology of acromegaly. Endocrine reviews. 1983 Summer; [PubMed PMID: 6354702]
Scacchi M,Cavagnini F, Acromegaly. Pituitary. 2006; [PubMed PMID: 17077948]
Jenkins PJ,Sohaib SA,Akker S,Phillips RR,Spillane K,Wass JA,Monson JP,Grossman AB,Besser GM,Reznek RH, The pathology of median neuropathy in acromegaly. Annals of internal medicine. 2000 Aug 1; [PubMed PMID: 10906834]
Nagulesparen M,Trickey R,Davies MJ,Jenkins JS, Muscle changes in acromegaly. British medical journal. 1976 Oct 16; [PubMed PMID: 974660]
Eugster E,Feingold KR,Anawalt B,Boyce A,Chrousos G,de Herder WW,Dhatariya K,Dungan K,Hershman JM,Hofland J,Kalra S,Kaltsas G,Koch C,Kopp P,Korbonits M,Kovacs CS,Kuohung W,Laferrère B,Levy M,McGee EA,McLachlan R,Morley JE,New M,Purnell J,Sahay R,Singer F,Sperling MA,Stratakis CA,Trence DL,Wilson DP, Gigantism Endotext. 2000 [PubMed PMID: 25905378]
Akirov A, Masri-Iraqi H, Dotan I, Shimon I. The Biochemical Diagnosis of Acromegaly. Journal of clinical medicine. 2021 Mar 9:10(5):. doi: 10.3390/jcm10051147. Epub 2021 Mar 9 [PubMed PMID: 33803429]
Level 2 (mid-level) evidenceMelmed S,Casanueva F,Cavagnini F,Chanson P,Frohman LA,Gaillard R,Ghigo E,Ho K,Jaquet P,Kleinberg D,Lamberts S,Laws E,Lombardi G,Sheppard MC,Thorner M,Vance ML,Wass JA,Giustina A, Consensus statement: medical management of acromegaly. European journal of endocrinology. 2005 Dec [PubMed PMID: 16322377]
Level 3 (low-level) evidenceFahlbusch R,Keller Bv,Ganslandt O,Kreutzer J,Nimsky C, Transsphenoidal surgery in acromegaly investigated by intraoperative high-field magnetic resonance imaging. European journal of endocrinology. 2005 Aug; [PubMed PMID: 16061830]
Abs R,Verhelst J,Maiter D,Van Acker K,Nobels F,Coolens JL,Mahler C,Beckers A, Cabergoline in the treatment of acromegaly: a study in 64 patients. The Journal of clinical endocrinology and metabolism. 1998 Feb; [PubMed PMID: 9467544]
Samson SL,Nachtigall LB,Fleseriu M,Gordon MB,Bolanowski M,Labadzhyan A,Ur E,Molitch M,Ludlam WH,Patou G,Haviv A,Biermasz N,Giustina A,Trainer PJ,Strasburger CJ,Kennedy L,Melmed S, Maintenance of Acromegaly Control in Patients Switching From Injectable Somatostatin Receptor Ligands to Oral Octreotide. The Journal of clinical endocrinology and metabolism. 2020 Oct 1 [PubMed PMID: 32882036]
Labadzhyan A,Nachtigall LB,Fleseriu M,Gordon MB,Molitch M,Kennedy L,Samson SL,Greenman Y,Biermasz N,Bolanowski M,Haviv A,Ludlam W,Patou G,Strasburger CJ, Oral octreotide capsules for the treatment of acromegaly: comparison of 2 phase 3 trial results. Pituitary. 2021 Dec [PubMed PMID: 34173129]
Jenkins PJ, The use of long-acting somatostatin analogues in acromegaly. Growth hormone & IGF research : official journal of the Growth Hormone Research Society and the International IGF Research Society. 2000 Apr [PubMed PMID: 10984265]
Level 1 (high-level) evidenceCaron P,Beckers A,Cullen DR,Goth MI,Gutt B,Laurberg P,Pico AM,Valimaki M,Zgliczynski W, Efficacy of the new long-acting formulation of lanreotide (lanreotide Autogel) in the management of acromegaly. The Journal of clinical endocrinology and metabolism. 2002 Jan [PubMed PMID: 11788630]
Cuevas-Ramos D,Fleseriu M, Pasireotide: a novel treatment for patients with acromegaly. Drug design, development and therapy. 2016 [PubMed PMID: 26811671]
Gadelha MR,Wildemberg LE,Kasuki L, The Future of Somatostatin Receptor Ligands in Acromegaly. The Journal of clinical endocrinology and metabolism. 2022 Jan 18; [PubMed PMID: 34618894]
Puig-Domingo M,Bernabéu I,Picó A,Biagetti B,Gil J,Alvarez-Escolá C,Jordà M,Marques-Pamies M,Soldevila B,Gálvez MA,Cámara R,Aller J,Lamas C,Marazuela M, Pasireotide in the Personalized Treatment of Acromegaly. Frontiers in endocrinology. 2021 [PubMed PMID: 33796079]
Trainer PJ,Drake WM,Katznelson L,Freda PU,Herman-Bonert V,van der Lely AJ,Dimaraki EV,Stewart PM,Friend KE,Vance ML,Besser GM,Scarlett JA,Thorner MO,Parkinson C,Klibanski A,Powell JS,Barkan AL,Sheppard MC,Malsonado M,Rose DR,Clemmons DR,Johannsson G,Bengtsson BA,Stavrou S,Kleinberg DL,Cook DM,Phillips LS,Bidlingmaier M,Strasburger CJ,Hackett S,Zib K,Bennett WF,Davis RJ, Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. The New England journal of medicine. 2000 Apr 20 [PubMed PMID: 10770982]
Level 1 (high-level) evidenceFleseriu M,Führer-Sakel D,van der Lely AJ,De Marinis L,Brue T,van der Lans-Bussemaker J,Hey-Hadavi J,Camacho-Hubner C,Wajnrajch MP,Valluri SR,Palladino AA,Gomez R,Salvatori R, More than a decade of real-world experience of pegvisomant for acromegaly: ACROSTUDY. European journal of endocrinology. 2021 Aug 27; [PubMed PMID: 34342594]
Chiloiro S,Bianchi A,Giampietro A,Pontecorvi A,Raverot G,Marinis L, Second line treatment of acromegaly: Pasireotide or Pegvisomant? Best practice & research. Clinical endocrinology & metabolism. 2022 Dec [PubMed PMID: 35931640]
Jenkins PJ,Bates P,Carson MN,Stewart PM,Wass JA, Conventional pituitary irradiation is effective in lowering serum growth hormone and insulin-like growth factor-I in patients with acromegaly. The Journal of clinical endocrinology and metabolism. 2006 Apr; [PubMed PMID: 16403824]
Level 2 (mid-level) evidenceLandolt AM,Haller D,Lomax N,Scheib S,Schubiger O,Siegfried J,Wellis G, Stereotactic radiosurgery for recurrent surgically treated acromegaly: comparison with fractionated radiotherapy. Journal of neurosurgery. 1998 Jun; [PubMed PMID: 9609294]
Geffner ME, The growth without growth hormone syndrome. Endocrinology and metabolism clinics of North America. 1996 Sep; [PubMed PMID: 8879991]
Dahlqvist P,Spencer R,Marques P,Dang MN,Glad CAM,Johannsson G,Korbonits M, Pseudoacromegaly: A Differential Diagnostic Problem for Acromegaly With a Genetic Solution. Journal of the Endocrine Society. 2017 Aug 1; [PubMed PMID: 29264563]
Sharma AN,Tan M,Amsterdam EA,Singh GD, Acromegalic cardiomyopathy: Epidemiology, diagnosis, and management. Clinical cardiology. 2018 Mar [PubMed PMID: 29574794]
[Problems in geriatric medicine]., Goto Y,, Nihon Shika Ishikai zasshi, 1978 [PubMed PMID: 25052731]
Level 3 (low-level) evidenceEsposito D,Ragnarsson O,Johannsson G,Olsson DS, Incidence of Benign and Malignant Tumors in Patients With Acromegaly Is Increased: A Nationwide Population-based Study. The Journal of clinical endocrinology and metabolism. 2021 Nov 19 [PubMed PMID: 34343297]
Melmed S,Casanueva FF,Klibanski A,Bronstein MD,Chanson P,Lamberts SW,Strasburger CJ,Wass JA,Giustina A, A consensus on the diagnosis and treatment of acromegaly complications. Pituitary. 2013 Sep; [PubMed PMID: 22903574]
Level 3 (low-level) evidenceUygur MM,Yazıcı DD,Buğdaycı O,Yavuz DG, Prevalence of vertebral fractures and serum sclerostin levels in acromegaly. Endocrine. 2021 Sep; [PubMed PMID: 34019235]
Silva PPB,Pereira RMR,Takayama L,Borba CG,Duarte FH,Trarbach EB,Martin RM,Bronstein MD,Tritos NA,Jallad RS, Impaired Bone Microarchitecture in Premenopausal Women With Acromegaly: The Possible Role of Wnt Signaling. The Journal of clinical endocrinology and metabolism. 2021 Aug 18 [PubMed PMID: 33871626]
Chiloiro S,Giampietro A,Gagliardi I,Bondanelli M,Veleno M,Ambrosio MR,Zatelli MC,Pontecorvi A,Giustina A,De Marinis L,Bianchi A, Impact of the diagnostic delay of acromegaly on bone health: data from a real life and long term follow-up experience. Pituitary. 2022 Dec; [PubMed PMID: 35922724]
Preo G,De Stefani A,Dassie F,Wennberg A,Vettor R,Maffei P,Gracco A,Bruno G, The role of the dentist and orthodontist in recognizing oro-facial manifestations of acromegaly: a questionnaire-based study. Pituitary. 2022 Feb [PubMed PMID: 34518997]
Broersen LHA,Zamanipoor Najafabadi AH,Pereira AM,Dekkers OM,van Furth WR,Biermasz NR, Improvement in Symptoms and Health-Related Quality of Life in Acromegaly Patients: A Systematic Review and Meta-Analysis. The Journal of clinical endocrinology and metabolism. 2021 Jan 23 [PubMed PMID: 33245343]
Level 2 (mid-level) evidenceColao A,Ferone D,Marzullo P,Lombardi G, Systemic complications of acromegaly: epidemiology, pathogenesis, and management. Endocrine reviews. 2004 Feb; [PubMed PMID: 14769829]
Abosch A,Tyrrell JB,Lamborn KR,Hannegan LT,Applebury CB,Wilson CB, Transsphenoidal microsurgery for growth hormone-secreting pituitary adenomas: initial outcome and long-term results. The Journal of clinical endocrinology and metabolism. 1998 Oct [PubMed PMID: 9768640]
Level 2 (mid-level) evidence