
Introduction
Episcleritis is an acute unilateral or bilateral inflammation of the episclera, the thin layer of tissue between the conjunctiva and sclera. The episclera is composed of loose connective tissue with its vascular supply coming from the anterior ciliary arteries, which are branches of the ophthalmic artery. Episcleritis can be diffuse, sectoral or nodular, and is most often idiopathic but is also often associated with systemic collagen vascular diseases, autoimmune diseases, and even some infections. Patient symptoms include redness, mild ocular discomfort or pain, and normal visual acuity. They rarely experience discharge or photophobia.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The majority of episcleritis cases are idiopathic, but 26% to 36% of patients have an associated systemic disorder that is responsible for the pathological process and development of episcleritis. These conditions include but are not limited to rheumatoid arthritis, Crohn disease, ulcerative colitis, psoriatic arthritis, systemic lupus erythematosus, reactive arthritis, relapsing polychondritis, ankylosing spondylitis, polyarteritis nodosa, Behcet disease, Cogan syndrome, and granulomatosis with polyangiitis, formerly called Wegener granulomatosis.
Some infections such as Lyme disease, cat scratch fever disease, syphilis, and those caused by the herpes virus are also linked to episcleritis but are much less common than the collagen vascular disease and autoimmune diseases listed above.[1][2][3][4][5]
Epidemiology
Episcleritis is most commonly diagnosed in young to middle-aged females and is rarely diagnosed in children. There is no consensus on general population incidence and prevalence because these studies are not published in the literature. However, it is known that diffuse episcleritis is significantly more prevalent than the nodular form of the condition. Diffuse episcleritis occurs in about 70% of patients; whereas, nodular episcleritis occurs in only about 30% of patients.[2][4]
It is also well established that the incidence and prevalence of episcleritis are higher in populations with systemic collagen-vascular disease and autoimmune diseases. In one study, recurrent episcleritis in the same or contralateral eye occurred in about 30% of patients.[1] The global statistics may likely be more as patients may self-diagnose and treat recurrent episodes.
Pathophysiology
The pathophysiology of episcleritis is a non-granulomatous inflammation of the episcleral vascular network.[1] This acute inflammatory process involves the activation of resident immune cells, including lymphocytes and macrophages. Once activated, they release inflammatory mediators causing vasodilation, increased vascular permeability, and the migration of more white blood cells and macrophages. The process is self-limited and generally lasts between 2 and 21 days.[1]
Histopathology
Histopathological studies are not often done due to the well known clinical features and relatively benign course of the disease. Under the microscope, the tissue will show the infiltration of lymphocytes, plasma cells, and macrophages along with edema. In a very small percentage of cases, episcleritis may be an initial manifestation of granulomatosis with polyangiitis. A histological sample of this tissue will look much different than one from a simple case of episcleritis. This tissue sample will include signs of granulomatous inflammation along with signs of collagen necrosis and vasculitis.[6][7] Similarly, a tissue sample from a patient with episcleritis associated with Cogan syndrome will also show signs of vasculitis.[8][9]
History and Physical
Episcleritis has an acute onset of sectoral or diffuse redness of one or both eyes. Episcleritis most often presents unilaterally, about 80% of the time, but may also have an acute bilateral presentation.[1] A nodule of inflamed tissue may be present but only in about 15% to 30% of cases. When present, the condition is named nodular episcleritis, and when absent, the condition is more appropriately named diffuse episcleritis. Patients will often describe tenderness or mild pain over the affected area but do not exhibit discharge, photophobia, or reduced visual acuity.
It may be necessary to determine the depth of the congested vessels to differentiate between episcleritis and scleritis. However, a patient with scleritis will be significantly more symptomatic with severe radiating pain, photophobia, tearing, and possibly reduced visual acuity. The clinician will instill one drop of 2.5% phenylephrine to the affected eye and evaluate the vasculature after 10 to 15 minutes. If the inflammation is localized to the episcleral tissue, then the vessels will blanch with the phenylephrine, and the eye will be relatively white and asymptomatic. Scleral vessels do not blanch with phenylephrine, and the eye will still appear to be significantly hyperemic if the scleral vessels are inflamed.
Patients with acute episcleritis are likely to have a concurrent ocular surface disease as well. Ocular rosacea is the most common, along with keratoconjunctivitis sicca and atopic keratoconjunctivitis.[1]
A review of systems should be completed, and if the patient has a history of or is currently suffering from joint/muscle pain or weakness, skin rash, psoriasis, diarrhea, oral or genital ulcers, history of drug abuse or possibility for sexually transmitted infections or any other symptoms to suggest a systemic association, then laboratory testing should be done.
Evaluation
When a systemic condition is suspected due to a positive review of systems, laboratory and radiographic testing should be completed. As discussed previously, these conditions may include rheumatoid arthritis, Crohn disease, ulcerative colitis, psoriatic arthritis, systemic lupus erythematosus, reactive arthritis, relapsing polychondritis, ankylosing spondylitis, polyarteritis nodosa, Behcet disease, Cogan syndrome, and granulomatosis with polyangiitis. Episcleritis may also be associated with Lyme disease, tuberculosis, syphilis, and herpes zoster.
Appropriate laboratory tests to rule out a systemic autoimmune inflammatory condition include a complete blood count with a differential, erythrocyte sedimentation rate/C reactive protein, rheumatoid factor, anti-nuclear antibody, anti-cyclic citrullinated peptide, HLA-B27, and anti-neutrophil cytoplasmic antibody (ANCA). If Lyme disease is suspected, then a Lyme antibody ELISA should be ordered. If tuberculosis (TB) is suspected, then a PPD skin test and a chest x-ray should be ordered. If syphilis is a consideration, then the patient should be tested with rapid plasma reagin (RPR) or VDRL and FTA-ABS or a treponemal-specific assay.
If the episcleral inflammation is persistent and does not respond to treatment (which will be discussed in the following section), then a tissue biopsy may be considered. While rare, a presentation of episcleritis may result from a systemic vasculitis such as granulomatosis with polyangiitis or Cogan syndrome, both of which can be fatal if not treated. Thus it is important to keep these rare instances in mind.[1]
Treatment / Management
Most cases of episcleritis are mild, transient, and will resolve without intervention within 2 to 21 days. Supportive treatment with refrigerated artificial tears at least four times daily is a common recommendation. Some patients require medical intervention depending on the severity of their symptoms. For those patients who require prescription medication, a mild topical corticosteroid such as fluorometholone 0.1% or loteprednol etabonate 0.5% may be prescribed four times a day for 1 to 2 weeks then tapered down. Although the risk of steroid response causing ocular hypertension is rare with these mild steroids, patients should followup 1 to 2 weeks after initiating treatment to monitor intraocular pressure and evaluate for resolution of the episcleritis. If the episcleral inflammation does not respond to fluorometholone 0.1% or loteprednol etabonate 0.5%, then the clinician may need to prescribe prednisolone acetate 1% four times a day. This is a more potent corticosteroid with greater anti-inflammatory effects but also has a higher risk of ocular hypertension. In general, the inflammation associated with episcleritis is not severe enough to warrant the use of difluprednate 0.05% or oral steroids. Topical steroids may also cause posterior sub-capsular cataracts and increase a patient’s susceptibility for infection; therefore, it is necessary that they are used judiciously.
Oral NSAIDs such as ibuprofen or naproxen may be used as an alternative to topical steroids or if topical steroids do not adequately resolve the inflammation. The dosage for ibuprofen is 200 to 600mg 3 to 4 times per day, and the dosage for naproxen is 250 to 500 mg twice per day for up to two weeks. Oral NSAIDs should be used with caution because of the risk of gastric ulcers and should be prescribed with an antacid such as omeprazole 20 mg daily or ranitidine 150 mg twice per day.
Topical NSAIDs such as diclofenac 0.1% and ketorolac 0.5% are of limited use, but some clinicians do prescribe these medications as a steroid alternative as well.[10] Topical NSAIDs may reduce the mild pain and inflammation associated with episcleritis without affecting intraocular pressure. Some older generic forms of diclofenac 0.1% were linked to corneal melt, and ketorolac 0.5% is known to cause significant stinging and burning upon instillation. Without a significant effect on the course of the disease, the benefits of topical NSAIDs do not outweigh the risk and disadvantages.(A1)
Differential Diagnosis
Misdiagnosis or delayed diagnosis of episcleritis is not common. The differential diagnosis list includes conditions that may appear to be similar to episcleritis, but after a thorough history and eye exam, misdiagnosis is uncommon.
Contact lens-associated red eye (CLARE) is a condition that may have a similar presentation to episcleritis but has a history and disease characteristics that make it difficult to mistake it for another condition. The patient affected by CLARE will have recently slept in their contact lenses and will have symptoms of unilateral pain, photophobia, and epiphora with normal visual acuity. The conjunctiva and cornea will show signs of inflammation, including corneal infiltrate and corneal edema and iritis, if severe.
A second condition on the differential diagnosis list should be acute conjunctivitis. Conjunctivitis is a broad term to include viral, bacterial, allergic/atopic, and toxic etiologies. Conjunctivitis most likely presents with an acute red eye with discharge, photophobia, itching/burning, and edematous eyelids. Depending on the specific cause, the patient will also exhibit conjunctival follicles, papillae, or a combination.
Sectoral conjunctival erythema and edema occur with phlyctenular conjunctivitis as well. This condition is caused by a delayed hypersensitivity reaction to antigens in the tear film and is often associated with blepharitis.
A mechanical inflammation of pinguecula causes pingueculitis. This is common when pingueculae are larger or co-occur with dry eye. Distinguishing features are that the inflamed area is associated with a pinguecula.
Iritis is another condition that may initially present similar to episcleritis, but the specific exam findings of iritis make it easy to differentiate. Patients with iritis will also have an acute onset of pain, redness, photophobia, and tearing. The conjunctival hyperemia is usually more concentrated in the circumlimbal area and is appropriately named ciliary flush. Distinguishing features of iritis are keratic precipitates on the corneal endothelium and cells and flare in the anterior chamber.
Scleritis is the most important condition to differentiate from episcleritis because the treatment of scleritis is more aggressive and can affect prognosis and complications. Patients with scleritis will complain of a more gradual onset of redness, pain, tearing, photophobia, and may have reduced visual acuity. These patients have deep, severe radiating pain from the affected eye, and their sclera will have a more red-purple hue. The hyperemia will not blanch with topical phenylephrine drops, and they may also have corneal involvement with peripheral stromal keratitis. More than 50% of patients with scleritis have a known systemic autoimmune connective tissue disease or vasculitis.[1] The treatment regimen for scleritis includes topical steroids and oral NSAIDs similar to episcleritis, but patients often need to be treated with oral steroids or subconjunctival steroid injections. Severe cases may require management with immunosuppressants with potentially devastating associated side effects such as azathioprine, methotrexate, and mycophenolate mofetil.
Prognosis
The prognosis of patients with episcleritis is generally good. Most of the patients do not have an underlying systemic condition, and while many patients will have recurrent episodes, the side effects of the inflammation and the treatments are rarely encountered and are manageable without significant intervention.
Enhancing Healthcare Team Outcomes
Episcleritis is an ocular condition in which the majority of patients who are affected have complete resolution of the inflammation and do not require ongoing management or follow up care with a specialist. However, 26% to 36% of patients with acute episcleritis will ultimately be diagnosed with systemic autoimmune disease and will need a referral to a specialist. Around 30% or more of patients will have recurrent episcleritis, and these patients require continuity of care with their primary eye care provider.[1][2][3][4][5]
Patients with an associated connective tissue disease or vasculitis should be referred to a rheumatologist specializing in their specific condition. The primary eye care provider is still included in the interprofessional care of the patient. Some patients with episcleritis will require topical steroids to manage their ocular inflammation, and if they are steroid responders, they may experience increased intraocular pressure. If the intraocular pressure cannot be managed appropriately by the primary eye care provider, then these patients will need to be referred to a glaucoma specialist for further intervention, possibly including surgery. Posterior sub-capsular cataracts can also occur in those patients with long-term topical corticosteroid use. This may require a referral to a cataract surgeon for cataract extraction and intraocular lens placement if cataracts become visually significant and affect the patient’s activities of daily living.
Although episcleritis is usually a benign condition with a good prognosis, there are some instances where there needs to be excellent interprofessional communication between the primary eye care provider and the rheumatologist, as well as between the primary eye care provider and the cataract surgeon or glaucoma specialist.
Media
(Click Image to Enlarge)
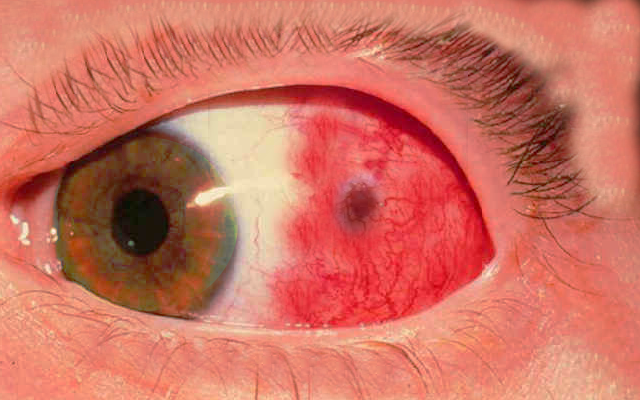
episcleritis of the eye
Image courtesy S BHimji MD
References
Akpek EK, Uy HS, Christen W, Gurdal C, Foster CS. Severity of episcleritis and systemic disease association. Ophthalmology. 1999 Apr:106(4):729-31 [PubMed PMID: 10201593]
Level 2 (mid-level) evidenceSainz de la Maza M, Molina N, Gonzalez-Gonzalez LA, Doctor PP, Tauber J, Foster CS. Clinical characteristics of a large cohort of patients with scleritis and episcleritis. Ophthalmology. 2012 Jan:119(1):43-50. doi: 10.1016/j.ophtha.2011.07.013. Epub 2011 Oct 2 [PubMed PMID: 21963265]
Level 2 (mid-level) evidenceDaniel Diaz J,Sobol EK,Gritz DC, Treatment and management of scleral disorders. Survey of ophthalmology. 2016 Nov - Dec [PubMed PMID: 27318032]
Level 3 (low-level) evidenceWatson PG, Hayreh SS. Scleritis and episcleritis. The British journal of ophthalmology. 1976 Mar:60(3):163-91 [PubMed PMID: 1268179]
Sainz de la Maza M, Jabbur NS, Foster CS. Severity of scleritis and episcleritis. Ophthalmology. 1994 Feb:101(2):389-96 [PubMed PMID: 8115160]
Kubaisi B, Abu Samra K, Foster CS. Granulomatosis with polyangiitis (Wegener's disease): An updated review of ocular disease manifestations. Intractable & rare diseases research. 2016 May:5(2):61-9. doi: 10.5582/irdr.2016.01014. Epub [PubMed PMID: 27195187]
Harper SL, Letko E, Samson CM, Zafirakis P, Sangwan V, Nguyen Q, Uy H, Baltatzis S, Foster CS. Wegener's granulomatosis: the relationship between ocular and systemic disease. The Journal of rheumatology. 2001 May:28(5):1025-32 [PubMed PMID: 11361183]
Level 2 (mid-level) evidenceD'Aguanno V,Ralli M,de Vincentiis M,Greco A, Optimal management of Cogan's syndrome: a multidisciplinary approach. Journal of multidisciplinary healthcare. 2018 [PubMed PMID: 29317827]
Grasland A,Pouchot J,Hachulla E,Blétry O,Papo T,Vinceneux P, Typical and atypical Cogan's syndrome: 32 cases and review of the literature. Rheumatology (Oxford, England). 2004 Aug [PubMed PMID: 15150435]
Level 3 (low-level) evidenceWilliams CP, Browning AC, Sleep TJ, Webber SK, McGill JI. A randomised, double-blind trial of topical ketorolac vs artificial tears for the treatment of episcleritis. Eye (London, England). 2005 Jul:19(7):739-42 [PubMed PMID: 15359265]
Level 1 (high-level) evidence