
Introduction
Dowling-Degos disease (DDD) is a rare autosomal dominant disorder, classically characterized by acquired reticular hyperpigmentation in flexural sites.[1] Onset is typically after puberty and commonly occurs in the third to fourth decade of life. Mutations in genes involved in melanosome trafficking and melanocyte and keratinocyte proliferation, differentiation, and cellular communications have all been implicated as etiologies. Hyperpigmentation is often recalcitrant to treatment, as evidenced by the varying success of topical and laser therapies.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Classic DDD and Galli-Galli disease (a variant of DDD) are caused by a loss-of-function mutation in the non-helical head domain of the keratin 5 gene (KRT5), leading to only one functional copy of KRT5.[2] Other genetic implications include mutations in POFUT1, POGLUT1, and PSENEN, which encode protein O-fucosyltransferase 1, protein O-glucosyltransferase 1, and presenilin enhancer protein 2 gene, respectively. POFUT1 and POGLUT1 mutations have been linked to a variant form of DDD that commonly involves non-flexural sites, while PSENEN mutations have been attributed to a variant of DDD associated with hidradenitis suppurativa.[3],[4]
Epidemiology
Since DDD was first described in 1938, there have been less than 50 cases reported in the literature. DDD has no racial or gender predilection, and lesions typically present in the third to fourth decade of life.[5]
Pathophysiology
Given that DDD is a disorder of hyperpigmentation, the main pathophysiology behind the disease fittingly involves melanosomal functioning. Intracellular transport, a function once believed to be reserved to microfilaments and microtubules, has been linked to keratins through studies elucidating the effect of haploinsufficiency of keratin 5 on the organization of cell adhesion, melanosome uptake into keratinocytes, organelle transport, and nuclear anchorage. Specifically, the head domain of keratin 5 is believed to play a role in melanosome trafficking through its interaction with Hsc70, a chaperone protein involved in vesicle uncoating.[2]
In addition, mutations in POFUT1 and POGLUT1 disrupt the Notch signaling pathway by affecting enzymes involved in post-translational modification. Mutations in PSENEN also affect the Notch signaling pathway by modifying an enhancer gene that encodes a component of y-secretase protein complex. The Notch signaling pathway plays an integral part in skin homeostasis by regulating the proliferation and differentiation of melanocytes and keratinocytes and mediating their interactions with one another.[3][4]
Histopathology
Histologically, DDD is characterized by an increase in pigmentation of the basal layer and downward elongation of the rete ridges with thinning of the underlying suprapapillary epithelium. This often appears filiform or antler-like when arising from the under surface of the epidermis and hair follicles. Dermal melanophages and a mild perivascular lymphohistiocytic infiltrate are also present.[5] Galli-Galli disease is a variant of DDD that has prominent suprabasal non-dyskeratotic acantholysis in addition to the classic histologic features previously described.[6] The importance of keratin 5 and Notch signaling in melanocyte and keratinocyte functioning is evidenced by the haphazard distribution of irregularly-shaped melanosomes and the irregular shape and size of keratinocytes on histopathology in patients with DDD.[3]
History and Physical
Patients with Dowling-Degos disease describe an abnormal darkening of the skin in their body folds and creases. Occasionally, patients report associated pruritus in these areas. On exam, reticulated hyperpigmentation is seen in intertriginous areas and is composed of lentigo-like brown macules and small brown papules with variable hyperkeratosis. The hyperpigmentation progressively increases over time, first arising in the axilla and groin and subsequently appearing in the intergluteal and inframammary folds, neck, trunk, and inner arms and thighs. [1][2][3][4][5]
Comedone-like lesions on the back and neck, pitted perioral scars, and rarely palmar pits may also be present, especially in patients with an overlap of classic DDD and reticulate acropigmentation of Kitamura.[7] Hidradenitis suppurative and epidermoid cysts have been described in a variant of DDD.[4] Rarely, lesions are located in non-flexural areas, as seen in those with mutations in the POFUT1 and POGLUT1 genes.[3] In addition, there has been a single case report of biopsy-confirmed DDD presenting as multiple hypopigmented macules in the classic flexural distribution, highlighting the evolving spectrum of DDD.[7]
Evaluation
DDD is a clinical and histopathologic diagnosis, confirmed by the history and physical exam and skin biopsy findings. Additional laboratory tests or imaging is not required for diagnosis.[5]
Treatment / Management
Topical hydroquinone, tretinoin, adapalene, and corticosteroids have been used with limited success. Case reports have shown success after treatment with Er:YAG laser followed by topical corticosteroids and fusidic acid. The Er:YAG laser is theorized to improve hyperpigmented areas by removing the abnormal proliferated epithelium and allowing formation of new epidermal tissue from unaffected follicular epithelium.[8][9][10](B3)
Differential Diagnosis
There are a variety of hyperpigmentation disorders that appear in the flexural areas that must be differentiated from DDD. They include the following:
- Reticulate acropigmentation of Kitamura
- Manifests as atrophic hyperpigmented papules that coalesce into a reticulated pattern. It is differentiated from DDD by its initial distribution on acral surfaces and onset in childhood.[11]
- Haber syndrome
- Initially presents as a photosensitive rosacea-like facial eruption during adolescence that is followed by keratotic papules, comedone-like lesions, pitted scars, and reticulate hyperpigmentation on the trunk, proximal extremities, and axillae. The initial facial eruption supports the classification of Haber's disease as a separate entity from DDD. [12][13]
- Neurofibromatosis type 1
- Exhibits axillary and inguinal freckling that mirrors the reticulated hyperpigmentation of DDD but becomes easily distinguishable from DDD with the development of multiple neurofibromas and other cutaneous manifestations.[14]
- Acanthosis nigricans
- Presents with velvety plaques that can be distinguished histologically from DDD by less elongation of the rete ridges and no follicular involvement.[15]
Enhancing Healthcare Team Outcomes
Dowling-Degos disease is a rare genodermatosis that afflicts a small number of the worldwide population. While there is no significant mortality associated with Dowling-Degos disease, patients may experience significant distress and anxiety due to the skin manifestations associated with this disease, especially those who exhibit acne-like scarring. This can lead to significant psychiatric morbidity and a decrease in quality of life. In addition, the minimal efficacy of treatments in Dowling-Degos disease and medication adverse effects may further precipitate psychological stress. Thus, an interprofessional approach with collaboration between dermatologists, psychiatrists, family medicine physicians, and pharmacists is recommended to improve patient outcomes. [Level 4 and 5][16]
Media
(Click Image to Enlarge)
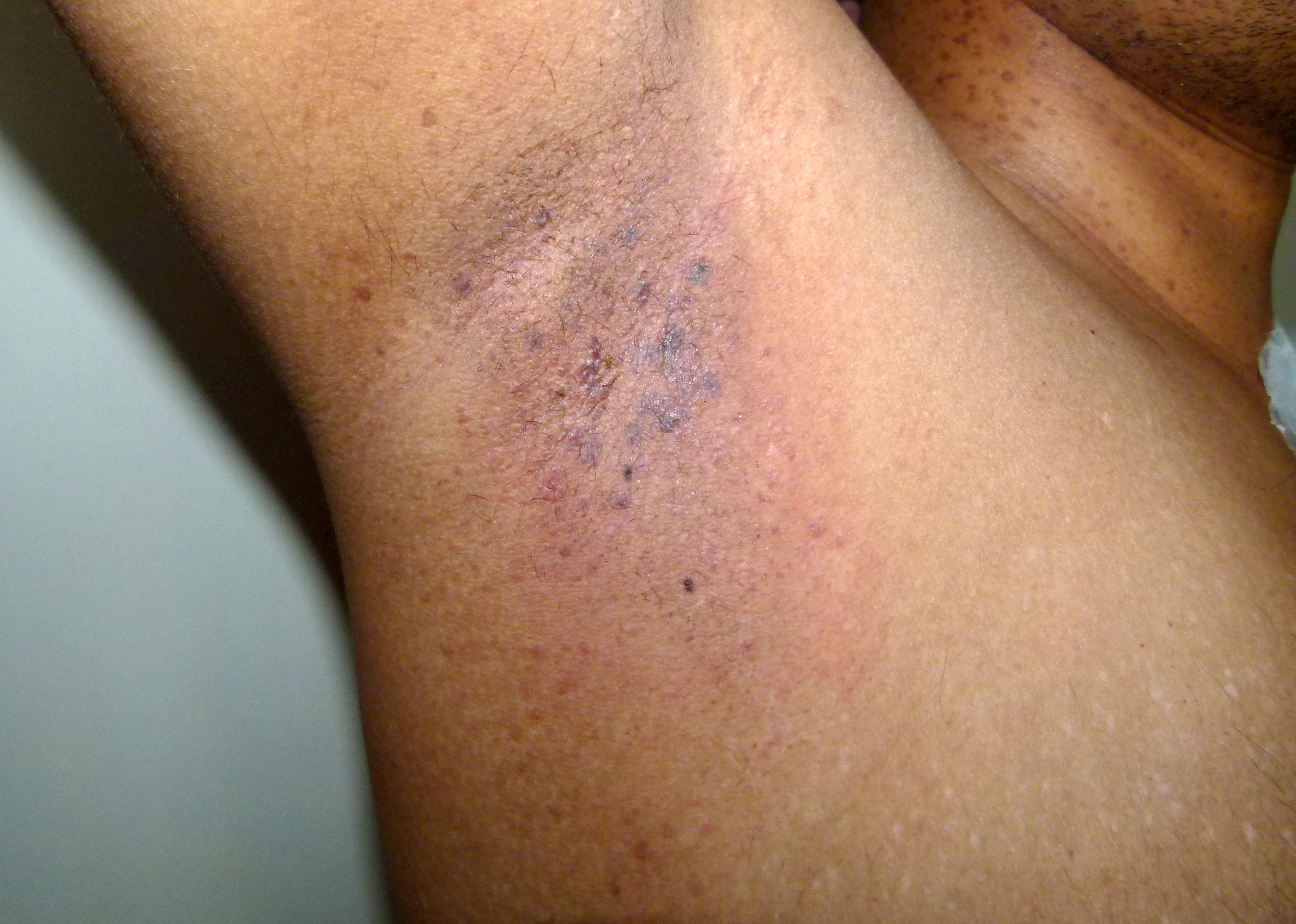
Dowling Degos Disease
Contributed by Dr. Shyam Verma, MBBS, DVD, FRCP, FAAD, Vadodara, India
References
Mahajan SH, Mahajan SA, Khopkar US, Kharkar VD. Follicular Dowling-Degos Disease: A Rare Pigmentary Dermatosis. Indian dermatology online journal. 2017 Nov-Dec:8(6):487-489. doi: 10.4103/idoj.IDOJ_311_16. Epub [PubMed PMID: 29204397]
Betz RC, Planko L, Eigelshoven S, Hanneken S, Pasternack SM, Bussow H, Van Den Bogaert K, Wenzel J, Braun-Falco M, Rutten A, Rogers MA, Ruzicka T, Nöthen MM, Magin TM, Kruse R. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. American journal of human genetics. 2006 Mar:78(3):510-9 [PubMed PMID: 16465624]
Basmanav FB, Oprisoreanu AM, Pasternack SM, Thiele H, Fritz G, Wenzel J, Größer L, Wehner M, Wolf S, Fagerberg C, Bygum A, Altmüller J, Rütten A, Parmentier L, El Shabrawi-Caelen L, Hafner C, Nürnberg P, Kruse R, Schoch S, Hanneken S, Betz RC. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. American journal of human genetics. 2014 Jan 2:94(1):135-43. doi: 10.1016/j.ajhg.2013.12.003. Epub [PubMed PMID: 24387993]
Ralser DJ, Basmanav FB, Tafazzoli A, Wititsuwannakul J, Delker S, Danda S, Thiele H, Wolf S, Busch M, Pulimood SA, Altmüller J, Nürnberg P, Lacombe D, Hillen U, Wenzel J, Frank J, Odermatt B, Betz RC. Mutations in γ-secretase subunit-encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. The Journal of clinical investigation. 2017 Apr 3:127(4):1485-1490. doi: 10.1172/JCI90667. Epub 2017 Mar 13 [PubMed PMID: 28287404]
Kim YC, Davis MD, Schanbacher CF, Su WP. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. Journal of the American Academy of Dermatology. 1999 Mar:40(3):462-7 [PubMed PMID: 10071319]
Level 2 (mid-level) evidenceHanneken S, Rütten A, Pasternack SM, Eigelshoven S, El Shabrawi-Caelen L, Wenzel J, Braun-Falco M, Ruzicka T, Nöthen MM, Kruse R, Betz RC. Systematic mutation screening of KRT5 supports the hypothesis that Galli-Galli disease is a variant of Dowling-Degos disease. The British journal of dermatology. 2010 Jul:163(1):197-200. doi: 10.1111/j.1365-2133.2010.09741.x. Epub 2010 Mar 5 [PubMed PMID: 20222933]
Level 1 (high-level) evidencePickup TL, Mutasim DF. Dowling-Degos disease presenting as hypopigmented macules. Journal of the American Academy of Dermatology. 2011 Jun:64(6):1224-5. doi: 10.1016/j.jaad.2009.11.015. Epub [PubMed PMID: 21571208]
Level 3 (low-level) evidenceYun JH, Kim JH, Choi JS, Roh JY, Lee JR. Treatment of Dowling-Degos disease with fractional Er:YAG laser. Journal of cosmetic and laser therapy : official publication of the European Society for Laser Dermatology. 2013 Dec:15(6):336-9. doi: 10.3109/14764172.2013.764437. Epub 2013 Mar 6 [PubMed PMID: 23464495]
Level 3 (low-level) evidenceWenzel J, Tappe K, Gerdsen R, Uerlich M, Philipp-Dormston W, Bieber T, Petrow W. Successful treatment of Dowling-Degos disease with Er:YAG laser. Dermatologic surgery : official publication for American Society for Dermatologic Surgery [et al.]. 2002 Aug:28(8):748-50 [PubMed PMID: 12174072]
Level 3 (low-level) evidenceWenzel G, Petrow W, Tappe K, Gerdsen R, Uerlich WP, Bieber T. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatologic surgery : official publication for American Society for Dermatologic Surgery [et al.]. 2003 Nov:29(11):1161-2 [PubMed PMID: 14641349]
Level 3 (low-level) evidenceAlshaikh H, Alsaif F, Aldukhi S. Clinical and Genetic Review of Hereditary Acral Reticulate Pigmentary Disorders. Dermatology research and practice. 2017:2017():3518568. doi: 10.1155/2017/3518568. Epub 2017 Oct 23 [PubMed PMID: 29201043]
McCormack CJ, Cowen P. Haber's syndrome. The Australasian journal of dermatology. 1997 May:38(2):82-4 [PubMed PMID: 9159964]
Level 3 (low-level) evidenceNishizawa A, Nakano H, Satoh T, Takayama K, Sawamura D, Yokozeki H. Haber's syndrome may be a clinical entity different from Dowling-Degos disease. The British journal of dermatology. 2009 Jan:160(1):215-7. doi: 10.1111/j.1365-2133.2008.08938.x. Epub 2008 Nov 28 [PubMed PMID: 19067693]
Level 3 (low-level) evidenceAdam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Friedman JM. Neurofibromatosis 1. GeneReviews(®). 1993:(): [PubMed PMID: 20301288]
Patel NU, Roach C, Alinia H, Huang WW, Feldman SR. Current treatment options for acanthosis nigricans. Clinical, cosmetic and investigational dermatology. 2018:11():407-413. doi: 10.2147/CCID.S137527. Epub 2018 Aug 7 [PubMed PMID: 30122971]
Basavaraj KH, Navya MA, Rashmi R. Relevance of psychiatry in dermatology: Present concepts. Indian journal of psychiatry. 2010 Jul:52(3):270-5. doi: 10.4103/0019-5545.70992. Epub [PubMed PMID: 21180416]