
Introduction
The immune system is the body’s defense mechanism against infections and is made of two pathways - the innate pathway and the adaptive pathway.[1] The innate pathway is present from birth and is pre-programmed. The innate immune system consists of the cellular component, which includes monocytes, macrophages, and natural killer cells, and the humoral component, which includes the complement system. The adaptive pathway develops during life with exposure to infection and increases affinity with experience. It consists of the cellular component, which is T cells, and the humoral component, which consists of antibodies made by B cells. The adaptive pathway facilitates improved destruction of the organism as well as forms memory cells for future infections.
Complement deficiencies are primary immunodeficiencies that cause various clinical scenarios depending on the specific complement protein that is deficient. This can include an increased risk of a wide range of infectious as well as local or systemic inflammatory and thrombotic conditions.[2]
Additionally, the complement system does not only act as the defense mechanism against infections it also works as a mediator in both the prevention of immune complex-related diseases and pathogenesis, such as systemic lupus erythematosus (SLE).[3][4][5]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Complement deficiencies are usually hereditary. The most common type is autosomal recessive form, including C1, C2, C3, C4, C5, C6, C7, C8, C9, and Mannan-binding lectin deficiencies.[2] The X-linked recessive pattern has been described for properdin deficiency.[6] Other complement deficiencies are acquired through complement overconsumption, protein synthesis dysfunction, protein loss disorders, autoimmunity, and high catabolic states. Deficiencies of early components of the classical complement pathway, including C1, C4, and C2, are associated with encapsulated bacterial infections like Streptococcus Pneumoniae and Haemophilus Influenza type b.[7] A deficiency of C3 is associated with severe recurrent pyogenic infections early in life.[8] Deficiencies of the late common pathway (C5, C6, C7, C8, and C9) are associated with increased Neisseria infections, including Neisseria meningitidis and Neisseria gonorrhoeae. A deficiency of properdin in the early alternative pathway is also associated with recurrent Neisseria infection.[9] A deficiency of mannan-binding lectin has been linked to an increased frequency of pyogenic infections and sepsis, particularly in children and neonates, when the adaptive immune system has not been matured. In addition to increased incidence of infection, an individual with early classical complement deficiency (C1, C4, or C2) frequently has a higher incidence of autoimmune disorders, especially systemic lupus erythematosus.[10] Early classical complements like C1 bind to cells undergoing apoptosis and facilitate the elimination of such cells. The body produces autoantibodies against these uncleared cells that result in autoimmune disorders.
Epidemiology
Complement deficiencies are rare worldwide; high-risk populations are screened to estimate prevalence. The mannan-binding lectin (MBL) of the lectin-based pathway is the most prevalent form of complement deficiency at 5% of the White population and it may be clinically silent. Apart from the MBL pathway, complement deficiencies are prevalent in 0.03% of the population.[11] Deficiency of C2 protein is the second most common form after MBL deficiency and is also clinically silent. C3 is a crucial player in the complement system as this protein is the last step of the early pathway, a precursor to C3a the anaphylatoxin as well as a facilitator of chemotaxis for neutrophils and macrophages. Among all patients with primary immunodeficiency diseases, approximately 5% have complement deficiency. The most common type of primary immunodeficiency is antibody deficiency consisting of approximately 65% of the cases.
In the cases of meningococcal infection, the prevalence rate is nearly 30%. Patients with C1q deficiency are observed to have a 93% chance of having SLE in the future. Similarly, C1rs deficiency is found to have a 57% association with SLE, and C4 deficiency is associated with a 75% association with SLE.
Properdin and C2 deficiencies have been more commonly observed in the White population, C6 deficiencies have been observed to have a predisposition in Africans, and deficiencies in C8 and C9 are more commonly seen in Asian populations. Specifically, two distinct C8 deficiency states have been studied: C8 alpha-gamma deficiency that is seen mostly in Afro-Caribbeans, Hispanics, and Japanese; and C8beta, mainly evident in Whites.
Pathophysiology
The complement system is a crucial part of the innate humoral immune system. The purpose of the complement system is to orchestrate opsonization, facilitate cytotoxic destruction and formulate membrane attack complexes (MAC), and the liberation of peptides that promote the inflammatory response.[12] The complement system consists of 3 pathways, the classical, alternative, and lectin, that are initiated by distinct mechanisms. The classical pathway is initiated by C1, C2, C4, and antigen-antibody immune complex. The lectin pathway is initiated by lectins, mannose-binding protein, sugar residues on the microbial surface, C4, and C2. The alternative pathway is initiated by C3, factor B, factor D, and properdin. These 3 pathways all share a common terminal pathway consisting of C5 to C9. The final outcome of the late pathway is to form a MAC that penetrates the cell membrane and facilitates cell death by lysis. Opsonization is the process of utilizing cleaved components of complement, C3b, and C4b, whereas inflammation is the process utilizing C3a and C5a, as anaphylatoxins.
Complement activity is regulated by an intricate system. The important components of this system include:
- Complement receptor 1 (CR1)
- Complement receptor 2 (CR2)
- Decay accelerating factor (DAF)
A North African study looking into the basis of complement factor I deficiency in cases of atypical hemolytic and uremic syndrome observed that the Ile357Met mutation could be a founding effect.[13]
There are cell surface-associated proteins and plasma proteins that regulate different steps of the complement pathways; for instance, factor H and factor I stop the formation of C3 convertase in the alternative pathway. The enzyme C1q esterase inhibits serine proteases C1r and C1s of the classic pathway. The deficiency of these regulatory proteins leads to the overactivation of the complement system and an abundance of damaging inflammatory effects.[14] Two clinical conditions due to these deficiencies are paroxysmal nocturnal hemoglobinuria (PNH) and hereditary angioedema. A sampling of thirteen patients of hereditary angioedema, mainly women younger than 50 years, with low or normal levels of C1 inhibitor who got COVID-19, had neither severe COVID-19 nor severe acute angioedema.[15]
History and Physical
The history is the most important initial diagnostic tool for ruling out complement immunodeficiency. Recurrent Neisseria infection indicates possible late complement deficiencies (C5-C9) and early alternative pathway (properdin) deficiency. Severe recurrent pyogenic infection early in life should be an indication to rule out C3 immunodeficiency.[16] Complement deficiency should also be suspected in a patient with recurrent sinopulmonary infection with normal humoral (antibody) immunity and with/without autoimmunity. Pyogenic infections and sepsis in children and neonates may be an indication of mannan-binding lectin deficiency.
Infants may develop Leiner disease, which presents as wasting, recurrent diarrhea, and generalized seborrheic dermatitis. It is attributed to a pathology of C5. However, there are reports suggesting a role of diminished C3 and reduced neutrophil mobility.[17][18] Thus, the etiology of Leiner disease is multifactorial.
There are three major pathophysiological sequelae of complement deficiencies:
Inadequate Opsonization
Opsonization is the process of making a pathogenic organism easy to ingest by the macrophage system. Defects in C3b or its cleavage product C3bi can lead to inadequate opsonization. The following can be various presentations of inadequate opsonization:
- Frequent sinopulmonary infections with pathogens such as Streptococcus pneumonia
- Autoimmune syndromes
- Encapsulated bacterial infections such as Neisseria meningitides and S pneumoniae
- Saccharomyces cerevisiae[19]
- Atypical hemolytic uremic syndrome
- Membranoproliferative glomerulonephritis
- Age-related macular degeneration
Defects in Cell Lysis
The defect in terminal cascade proteins also predisposes individuals to infection; however, the clinical history in these cases is different. The following are some consequences of these defects:
- Infection at an older age as opposed to patients with other complement deficiencies
- Lack of protection against many other nonbacterial pathogens, such as viruses, fungi, and mycobacteria
Immune Complex Diseases
Patients with the classic pathway complement deficiencies are more prone to develop immune complex diseases. The following are some sequelae of such defects:
- Seizures secondary to neuropsychiatric systemic lupus erythematosus[20]
- Antiphospholipid antibody syndrome
- Arterial thrombosis[21]
The examination may be normal if the patient is not actively infected or has autoimmunity. In a patient with active pneumococcal pneumonia, a physical exam demonstrates crackles or bronchial breath sounds on lung auscultation at peripheral lung fields. Egophony can be noted when significant consolidation is present. Examination of patients with meningitis frequently reveals meningismus. Patients may have positive Kernig or Brudzinski signs that show meningeal irritation. Other signs of Neisseria meningitidis infection include petechial rash and, in severe cases, can present with septic shock or disseminated intravascular coagulation (DIC).[22] Patients with early classical complement deficiencies may present with swollen and/or inflamed joints, suggesting autoimmunity.
Evaluation
Patients with recurrent infections of the respiratory tract without risk factors (negative human immunodeficiency virus, no asplenia, no other immunodeficiencies), as well as recurrent infections of encapsulated organisms such as Streptococcus pneumoniae, Neisseria meningitidis, and Hemophilus influenzae, should be screened for complement deficiency.[23] Patients with family members who have the recurrent pneumococcal and meningococcal disease should also be screened.
Screening for the complement system includes tests for the classical complement pathway (CH50), the alternate pathway (AH50), and the mannose-binding lectin (MBL) pathway (MBL by ELISA). Low CH50 and normal AH50 suggest early classical complement component (C1, C2, and C4) deficiency. Low AH50 with normal CH50 suggests a deficiency of early alternative complement pathway factors (factor B, factor D, and properdin). Low AH50 and low CH50 suggest common terminal complement (C3, C5, C6, C7, C8, or C9) deficiency. If CH50 and AH50 are both normal and the clinician still suspects complement deficiency, MBL functional assay is indicated to screen for MBL deficiency.[24]
Consider performing a head CT scan before a lumbar puncture in a patient suspected to have meningitis. Individuals with classic complement pathway deficiencies must be screened for consequences of immune complex diseases with the help of urinalysis and a complete blood cell count. A lumbar puncture should be performed in cases suspected of having possible meningitis to diagnose bacterial meningitis.
Treatment / Management
Complement deficiency is not typically treated with complement protein supplementation due to the infeasibility of overly frequent infusions, the risk for bloodborne infections, and the risk of developing antibodies to exogenous complement proteins. This condition is managed on a case-by-case basis with antibiotics for each episode of infection as well as regular visits with their immunologist.[25] Due to this population’s vulnerability to encapsulated organisms; patients need to be aware of the symptoms of meningococcal infection and seek care immediately if they should develop. Patients with complement deficiency should undergo all routine bacterial and viral vaccinations, especially meningococcal and pneumococcal vaccinations, and do not have a contraindication to live vaccines.[26](B3)
Some patients may develop flare-ups of their autoimmune disease. These patients should be managed with immunosuppressive therapy.
Differential Diagnosis
The differential diagnosis for these recurrent infections broadly includes B cell immunodeficiency, combined immunodeficiency, acquired immunodeficiencies, as well as asplenia with a predisposition for encapsulated organisms. The list of differential diagnoses is:
- Asplenia
- Common variable deficiency
- Hypogammaglobulinemia
- HIV
- Chronic granulomatous disease
- Chediak-Higashi syndrome
- Leukocyte adhesion deficiency
- Cyclic neutropenia
- SLE[27]
- Immunoglobulin A deficiency
- Immunoglobulin G deficiency
- Immunoglobulin D deficiency
- Immunoglobulin M deficiency
- Meningococcemia
Prognosis
The prognosis of this condition depends on the recurrence of infections as well as the severity of the episode of infection at the time. Many of these patients are at high risk for meningitis which can be life-threatening if untreated.[22]
Complications
Complications include severe pneumococcal and meningococcal infections that can be fatal if not evaluated and treated adequately. Patients may also develop autoimmune disorders, especially systemic lupus erythematosus.
Consultations
Patients with recurrent pneumococcal and/or meningococcal infections without predisposing risk factors should be referred to the allergist/immunologist. Also, consider consulting an infectious disease specialist or a rheumatologist to help treat acute complications of the complement deficiencies.
Deterrence and Patient Education
Patients and parents should be educated on the symptoms of serious illness, including meningitis, pneumonia, and sepsis. They should be instructed to seek care immediately if any of these signs develop. They should also seek care early for milder infections so that appropriate antibiotics can be started, and patients may be given prophylactic antibiotics for sudden recurrences. Vaccination is an important preventive measure, especially vaccination to prevent meningococcal and pneumococcal infections.
Relatives of patients with complement deficiency should undergo complement screening as a diagnosis may allow prophylaxis to prevent possible disabling or life-threatening infections.[28] Complement deficiency should be looked for in adults with N meningitidis infection.[23] Early diagnosis, antibiotic prophylaxis, and the use of vaccinations may allow normal life in hereditary C2 deficiency.[29][7]
Enhancing Healthcare Team Outcomes
Complement deficiency can lead to life-threatening infections as well as long-term autoimmune conditions and organ injuries. The interprofessional team of the primary care clinician and emergency medicine clinician must be aware of the clinical features of patients with complement deficiency or immunodeficiency in general. Relying on acute treatment when the patient is acutely ill may not be adequate to improve the patient's quality of life and clinical outcome. A high index of suspicion is important, and early referral to allergist/immunologists may be important when the diagnosis of immunodeficiency is not clear.
Media
(Click Image to Enlarge)
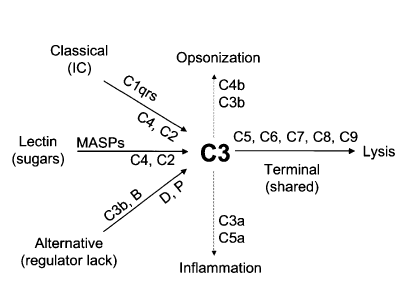
Complement cascade schematic diagram
Contributed by Lena Wen
References
Müller-Eberhard HJ. Complement. Annual review of biochemistry. 1975:44():697-724 [PubMed PMID: 1094920]
Schröder-Braunstein J, Kirschfink M. Complement deficiencies and dysregulation: Pathophysiological consequences, modern analysis, and clinical management. Molecular immunology. 2019 Oct:114():299-311. doi: 10.1016/j.molimm.2019.08.002. Epub 2019 Aug 14 [PubMed PMID: 31421540]
Blanco P, Pellegrin JL, Moreau JF, Viallard JF. [Pathophysiology of systemic lupus erythematosus]. Presse medicale (Paris, France : 1983). 2007 May:36(5 Pt 2):825-34 [PubMed PMID: 17449371]
Level 3 (low-level) evidenceContin-Bordes C, Lazaro E, Pellegrin JL, Viallard JF, Moreau JF, Blanco P. [Systemic lupus erythematosus: from pathophysiology to treatment]. La Revue de medecine interne. 2009 Dec:30(12 Suppl):H9-13 [PubMed PMID: 19995652]
Level 3 (low-level) evidenceSkattum L, van Deuren M, van der Poll T, Truedsson L. Complement deficiency states and associated infections. Molecular immunology. 2011 Aug:48(14):1643-55. doi: 10.1016/j.molimm.2011.05.001. Epub 2011 May 31 [PubMed PMID: 21624663]
Level 3 (low-level) evidenceLee JX, Yusin JS, Randhawa I. Properdin deficiency-associated bronchiectasis. Annals of allergy, asthma & immunology : official publication of the American College of Allergy, Asthma, & Immunology. 2014 Jun:112(6):557-9. doi: 10.1016/j.anai.2014.04.003. Epub 2014 May 1 [PubMed PMID: 24793003]
Level 3 (low-level) evidenceJönsson G, Hansson C, Mellhammar L, Gullstrand B, Bengtsson AA, Sahl C, Skattum L. Increased serum bactericidal activity of autologous serum in C2 deficiency after vaccination against Haemophilus influenzae type b, and further support for an MBL-dependent C2 bypass mechanism. Vaccine. 2021 Feb 22:39(8):1297-1302. doi: 10.1016/j.vaccine.2021.01.007. Epub 2021 Jan 25 [PubMed PMID: 33509693]
Miller EC, Chase NM, Densen P, Hintermeyer MK, Casper JT, Atkinson JP. Autoantibody stabilization of the classical pathway C3 convertase leading to C3 deficiency and Neisserial sepsis: C4 nephritic factor revisited. Clinical immunology (Orlando, Fla.). 2012 Dec:145(3):241-50. doi: 10.1016/j.clim.2012.09.007. Epub 2012 Sep 28 [PubMed PMID: 23117396]
Level 3 (low-level) evidenceAgarwal S, Ferreira VP, Cortes C, Pangburn MK, Rice PA, Ram S. An evaluation of the role of properdin in alternative pathway activation on Neisseria meningitidis and Neisseria gonorrhoeae. Journal of immunology (Baltimore, Md. : 1950). 2010 Jul 1:185(1):507-16. doi: 10.4049/jimmunol.0903598. Epub 2010 Jun 7 [PubMed PMID: 20530262]
Bryan AR, Wu EY. Complement deficiencies in systemic lupus erythematosus. Current allergy and asthma reports. 2014 Jul:14(7):448. doi: 10.1007/s11882-014-0448-2. Epub [PubMed PMID: 24816552]
Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Molecular immunology. 2014 Oct:61(2):110-7. doi: 10.1016/j.molimm.2014.06.030. Epub 2014 Jul 15 [PubMed PMID: 25037634]
Level 3 (low-level) evidenceGigli I. Immunochemistry and immunobiology of the complement system. The Journal of investigative dermatology. 1976 Sep:67(3):346-53 [PubMed PMID: 787430]
Jlajla H, Dehman F, Jallouli M, Khedher R, Ayadi I, Zerzeri Y, Laadhar L, Sfar I, Mahfoudh A, Gorgi Y, Cheour E, Zouaghi K, Gargah T, Kallel Sellami M. Molecular basis of complement factor I deficiency in Tunisian atypical haemolytic and uraemic syndrome patients. Nephrology (Carlton, Vic.). 2019 Mar:24(3):357-364. doi: 10.1111/nep.13217. Epub [PubMed PMID: 29292855]
Roumenina LT, Sène D, Radanova M, Blouin J, Halbwachs-Mecarelli L, Dragon-Durey MA, Fridman WH, Fremeaux-Bacchi V. Functional complement C1q abnormality leads to impaired immune complexes and apoptotic cell clearance. Journal of immunology (Baltimore, Md. : 1950). 2011 Oct 15:187(8):4369-73. doi: 10.4049/jimmunol.1101749. Epub 2011 Sep 19 [PubMed PMID: 21930969]
Level 3 (low-level) evidenceGrumach AS, Goudouris E, Dortas Junior S, Marcelino FC, Alonso MLO, Martins RO, Arpon MA, Valle SOR. COVID-19 affecting hereditary angioedema patients with and without C1 inhibitor deficiency. The journal of allergy and clinical immunology. In practice. 2021 Jan:9(1):508-510. doi: 10.1016/j.jaip.2020.11.042. Epub 2020 Nov 30 [PubMed PMID: 33271349]
Schur PH. Inherited complement component abnormalities. Annual review of medicine. 1986:37():333-46 [PubMed PMID: 3010807]
Sonea MJ, Moroz BE, Reece ER. Leiner's disease associated with diminished third component of complement. Pediatric dermatology. 1987 Aug:4(2):105-7 [PubMed PMID: 2958789]
Level 3 (low-level) evidenceGoodyear HM, Harper JI. Leiner's disease associated with metabolic acidosis. Clinical and experimental dermatology. 1989 Sep:14(5):364-6 [PubMed PMID: 2532987]
Level 3 (low-level) evidenceDegn SE, Jensenius JC, Thiel S. Disease-causing mutations in genes of the complement system. American journal of human genetics. 2011 Jun 10:88(6):689-705. doi: 10.1016/j.ajhg.2011.05.011. Epub [PubMed PMID: 21664996]
Level 3 (low-level) evidencevan Schaarenburg RA, Magro-Checa C, Bakker JA, Teng YK, Bajema IM, Huizinga TW, Steup-Beekman GM, Trouw LA. C1q Deficiency and Neuropsychiatric Systemic Lupus Erythematosus. Frontiers in immunology. 2016:7():647. doi: 10.3389/fimmu.2016.00647. Epub 2016 Dec 27 [PubMed PMID: 28082982]
Peerschke EI, Yin W, Alpert DR, Roubey RA, Salmon JE, Ghebrehiwet B. Serum complement activation on heterologous platelets is associated with arterial thrombosis in patients with systemic lupus erythematosus and antiphospholipid antibodies. Lupus. 2009 May:18(6):530-8. doi: 10.1177/0961203308099974. Epub [PubMed PMID: 19395455]
Level 2 (mid-level) evidenceLewis LA, Ram S. Meningococcal disease and the complement system. Virulence. 2014 Jan 1:5(1):98-126. doi: 10.4161/viru.26515. Epub 2013 Oct 8 [PubMed PMID: 24104403]
Audemard-Verger A, Descloux E, Ponard D, Deroux A, Fantin B, Fieschi C, John M, Bouldouyre A, Karkowsi L, Moulis G, Auvinet H, Valla F, Lechiche C, Davido B, Martinot M, Biron C, Lucht F, Asseray N, Froissart A, Buzelé R, Perlat A, Boutboul D, Fremeaux-Bacchi V, Isnard S, Bienvenu B. Infections Revealing Complement Deficiency in Adults: A French Nationwide Study Enrolling 41 Patients. Medicine. 2016 May:95(19):e3548. doi: 10.1097/MD.0000000000003548. Epub [PubMed PMID: 27175654]
Ekdahl KN, Persson B, Mohlin C, Sandholm K, Skattum L, Nilsson B. Interpretation of Serological Complement Biomarkers in Disease. Frontiers in immunology. 2018:9():2237. doi: 10.3389/fimmu.2018.02237. Epub 2018 Oct 24 [PubMed PMID: 30405598]
Ram S, Lewis LA, Rice PA. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clinical microbiology reviews. 2010 Oct:23(4):740-80. doi: 10.1128/CMR.00048-09. Epub [PubMed PMID: 20930072]
Level 3 (low-level) evidenceSobh A, Bonilla FA. Vaccination in Primary Immunodeficiency Disorders. The journal of allergy and clinical immunology. In practice. 2016 Nov-Dec:4(6):1066-1075. doi: 10.1016/j.jaip.2016.09.012. Epub [PubMed PMID: 27836056]
Műzes G, Sipos F. [Primary immunodeficiency and autoimmune diseases]. Orvosi hetilap. 2018 Jun:159(23):908-918. doi: 10.1556/650.2018.31064. Epub [PubMed PMID: 29860882]
Dellepiane RM,Dell'Era L,Pavesi P,Macor P,Giordano M,De Maso L,Pietrogrande MC,Cugno M, Invasive meningococcal disease in three siblings with hereditary deficiency of the 8(th) component of complement: evidence for the importance of an early diagnosis. Orphanet journal of rare diseases. 2016 May 17 [PubMed PMID: 27183977]
Dellepiane RM, Baselli LA, Cazzaniga M, Lougaris V, Macor P, Giordano M, Gualtierotti R, Cugno M. Hereditary Deficiency of the Second Component of Complement: Early Diagnosis and 21-Year Follow-Up of a Family. Medicina (Kaunas, Lithuania). 2020 Mar 10:56(3):. doi: 10.3390/medicina56030120. Epub 2020 Mar 10 [PubMed PMID: 32164349]