
Introduction
Bullous pemphigoid (BP) is the most common autoimmune subepidermal blistering disorder, representing 80% of subepidermal immunobullous cases.[1] Bullous pemphigoid most commonly affects elderly patients between the ages of 60 to 80 years. While the clinical presentation of bullous pemphigoid is broad, the immunobullous skin disorder characteristically presents with tense bullae and intense generalized pruritus. In atypical cases, bullous lesions may be absent, and these cases require a high degree of clinical suspicion. A biopsy for hematoxylin and eosin staining will show a subepidermal split with eosinophils, and direct immunofluorescence will highlight the autoantibodies against the basement membrane zone. ELISA testing is also useful in diagnosing bullous pemphigoid. Treatment depends on the severity of the disease; however, standard therapies involve topical or systemic immunosuppressive agents.[2] Prognosis varies, and long-term monitoring is often required.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
While most bullous pemphigoid cases are due to autoantibodies against proteins arranged at the dermal-epidermal junction, some cases of bullous pemphigoid are caused by systemic medications. Drug-induced bullous pemphigoid occurs up to three months after medication initiation and is typically noted in a younger subset of patients. The drugs reported in the literature to cause bullous pemphigoid-like eruptions to include diuretics such as furosemide and spironolactone, NSAIDs, amoxicillin, PD-1/PD-L1 inhibitors, gliptins, and TNF-alpha inhibitors.[3]
Epidemiology
As mentioned above, BP affects individuals older than 60 years of age.[4] Annual incidence is 6 to 13 new cases per million people in the United States, while it affects 12 to 13 per million in Central Europe. It has an equal incidence in both males and females and has no racial bias. This disease may present in childhood, but this is rare. Specific HLA class II alleles can be found in BP patients, including the allele DQB1*0301 in White race patients, and alleles DRB1*04, DRB1*1101, and DQB1*0302 in Japanese patients.[4][5]
Pathophysiology
There are 2 main components to the pathophysiology of BP: immunologic and inflammatory. The immunologic elements comprise autoantibodies against 2 parts of the basal keratinocyte hemidesmosomal proteins BP antigen 230 (BPAG1) and BP antigen 180 (BPAG2 or type XVII collagen).[6] These antigens play an essential part in the adhesion complexes that promote epithelial-stromal adhesion. When autoantibodies bind to their target antigen, the inflammatory component follows, activating complement and mast cells. This causes neutrophils and eosinophils to release various inflammatory cells resulting in the release of proteolytic enzymes that damage the dermal-epidermal junction.[7]
Histopathology
Histopathology will reveal a subepidermal split with a superficial perivascular inflammatory infiltrate and numerous eosinophils. Spongiosis and superficial papillary dermal infiltrate of eosinophils without vesiculation are characteristic features of urticarial lesions and may indicate the dermatopathologist to a diagnosis of urticarial or pre-bullous pemphigoid.[8] In these cases, direct immunofluorescence studies are imperative.
Direct immunofluorescence involves directly detecting tissue bound autoantibodies and is the gold standard of evaluation in autoimmune blistering diseases. It is imperative to biopsy the site of the skin lesion to get direct immunofluorescence. At least 2 punch biopsies should be obtained at the time of evaluation: one to send for hematoxylin and eosin staining, and the other is perilesional, uninvolved skin for direct immunofluorescence. DIF pattern for bullous pemphigoid will show deposition of C3 and IgG in a linear homogeneous pattern at the basement membrane zone.[9] In the early stages of this skin disease, only C3 may be present.[10] A salt-split skin immunofluorescence study may be done where the patient serum is applied on the salt-split skin, which splits the skin at the level of the lamina lucida. Bullous pemphigoid immunoreactants will localize to the epidermal side of the preparation, versus epidermolysis bullosa acquisita, which localizes to the dermal side.[11][12]
History and Physical
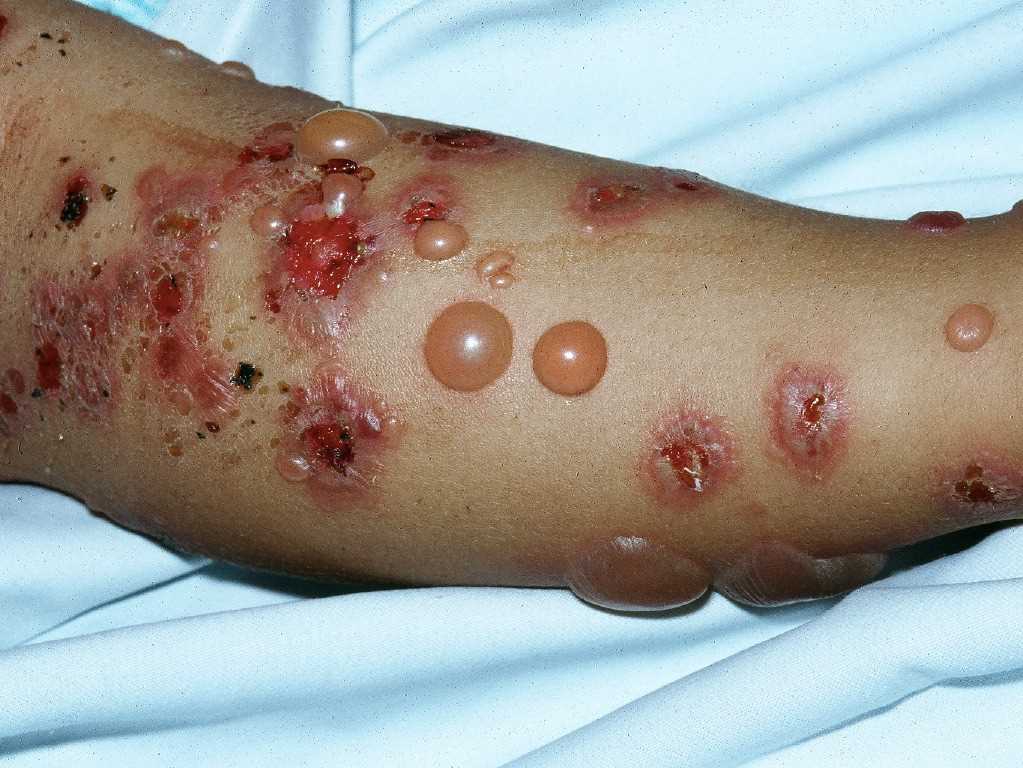
In the prodromal phase, patients may experience moderate-to-severe pruritus alone or associated with urticarial, papular lesions.[13] This later evolves to bullae in weeks to months, typically present in the axillae, on the flexor surface of the forearms, medial thighs, trunk, and abdomen. Approximately 20% of patients will have neither bullae nor erosions at the time of presentation.[14] Constitutional symptoms are uncommon, except in widespread, severe disease. The bullous phase characteristically presents with vesicles and bullae on a background of normal or erythematous skin. The blisters are tense, up to 1 to 4 centimeters in diameter, and sometimes hemorrhagic. They typically contain clear fluid and may persist for several days before leaving erosions and crusts. Unlike pemphigus vulgaris, the Nikolsky sign is negative in typical cases of bullous pemphigoid (see Image. Bullous Pemphigoid).
Childhood cases are rare. However, there have been cases of infantile bullous pemphigoid, which presents in an acral distribution in the first year of life. [15][16] The other variant of bullous pemphigoid of childhood are cases localized to the vulva.[17] These cases need to be distinguished from other disorders due to the rarity of the former. Therefore, direct immunofluorescence may be necessary. In some childhood cases, IgA antibodies against the NC16A domain of BP180 have been noted.[18] It is also reported that the infancy subtype is not related to a maternofetal transfer of autoantibodies.[19]
Researchers debate whether bullous pemphigoid is associated with internal malignancy. However, most believe this association is due to the older age of the patient. A recent study found no increased risk for concurrent or subsequent malignancies in patients with bullous pemphigoid.[20]
Evaluation
Diagnosis of the bullous pemphigoid relies on the clinical scenario and laboratory tests. Histology with direct and indirect immunofluorescence studies aid in diagnosis.
Histology will show a subepidermal split with a superficial perivascular inflammatory infiltrate and numerous eosinophils. Spongiosis and superficial papillary dermal infiltrate of eosinophils without vesiculation characterize urticarial lesions and may provide a clue to the dermatopathologist for diagnosing urticarial or pre-bullous pemphigoid.[8] Direct immunofluorescence studies are necessary.
Direct immunofluorescence involves directly detecting tissue bound autoantibodies and is the gold standard of evaluation in autoimmune blistering diseases. It is necessary to biopsy the site of the skin lesion for direct immunofluorescence. At least two punch biopsies should be obtained at the time of evaluation: one for hematoxylin and eosin staining and another using perilesional, uninvolved skin for direct immunofluorescence. DIF pattern for bullous pemphigoid demonstrates deposition of C3 and IgG in a linear homogeneous pattern at the basement membrane zone.[9] In the early stages of skin disease, only C3 may be present.[10] A salt-split skin immunofluorescence study is an option, where the patient serum is applied on salt-split skin, which splits the skin at the level of the lamina lucida. Bullous pemphigoid immunoreactants will localize to the epidermal side of the preparation, versus epidermolysis bullosa acquisita, which localizes to the dermal side.[11][12]
ELISA testing to detect antibodies to the NC16A domain of BP180, also known as BPAG2, is available and has a sensitivity of 89% and specificity of 98%. Autoantibodies to BP180 and BP230 may be identified in normal subjects without bullous pemphigoid, but they will not bind to the NC16A domain. [21][22]
The clinical predictors of bullous pemphigoid were described in a report from the French Bullous Study Group in 1998. They suggested that patients with a subepidermal blistering disorder associated with linear deposits of IgG or C3 along the epidermal basement membrane without skin atrophy, mucosal or head, and neck involvement, and older than 70 years strongly supports a diagnosis of BP. Their reports stated three out of four diagnostic criteria have a sensitivity of 90% and specificity of 83%.[1]
Treatment / Management
The mainstay of treatment for bullous pemphigoid is systemic corticosteroids, but treatment ultimately depends on comorbidities and the extent of the disease. For localized disease, less than 20% body surface area in an elderly patient, super-potent topical steroids such as clobetasol may be used.[23] Topical steroids combined with nicotinamide plus tetracycline, minocycline, or doxycycline have shown success in multiple cases.[24](A1)
For extensive disease, systemic prednisone at a dose of 0.5 to 1.0mg/kg per day is recommended. This dose of systemic corticosteroids controls the disease within approximately two weeks and can be slowly tapered over six to nine months or longer. This treatment regimen is limited by patient age, comorbidities, and side effects. Potent topical corticosteroids can control generalized BP with fewer systemic side effects.[25](B3)
Immunosuppressive therapy is used when steroids do not control the disease or if patients have contraindications for systemic corticosteroid treatments. Alternative agents include azathioprine, mycophenolate mofetil, methotrexate, chlorambucil, and cyclophosphamide. If all other treatments fail, IVIG, anti-CD20 (rituximab), or omalizumab can be used for treatment-resistant cases.[25](B3)
Serum IgG autoantibodies levels to BP180 have been shown to correlate with disease severity in several ELISA-based studies. Also, a high BP180-NC16A ELISA score and positive direct immunofluorescence at the end of therapy show possible relapse.[14][26](B2)
Differential Diagnosis
The presence of bullae can be a nonspecific finding and resemble various dermatoses depending on the timing in the clinical course. Drug reactions, contact dermatitis, urticarial dermatoses, arthropod reactions, other autoimmune bullous disorders such as pemphigus vulgaris, and scabies. Bullae can also form secondary to allergic contact dermatitis, Stevens-Johnson syndrome, dyshidrotic eczema, pseudoporphyria, or porphyria cutanea tarda. If bullae are identified in childhood, bullous impetigo, epidermolysis bullosa, and bullous variants of mastocytosis should be considered in the differential.[14]
Histopathology, direct and indirect immunofluorescence studies, salt-split skin, and ELISA testing may be necessary if the diagnosis of bullous pemphigoid is questioned.
Prognosis
Bullous pemphigoid typically resolves spontaneously in a few months but can remain for up to 5 years. Treatment, as outlined above, can provide symptomatic relief of pain and itching.[27]
Complications
Morbidity and mortality of bullous pemphigoid can include:
- Staphylococcal and streptococcal skin infections and sepsis
- Viral infections, including herpes simplex or varicella-zoster
- Adverse effects of treatment
Deterrence and Patient Education
This disease can render the patient's skin fragile and susceptible to microbial invasion. Patients need to avoid trauma to their skin. Skin hygiene is a crucial component of preventing complications, and patients need to recognize both complications and adverse events from treatment so they can report these to their clinician. They also need to understand the gravity of the condition.
Enhancing Healthcare Team Outcomes
Most general practitioners, nurse practitioners, and primary care providers may not be familiar with BP. This is a serious skin disorder with a very high morbidity and mortality rate. When patients present with large bullae, it is important to seek a dermatology consultation. The sooner the disorder is treated, the better the prognosis.
Bullous pemphigoid is typically a chronic disease with unpredictable exacerbations, with up to 30% of affected patients relapsing within the first year. When treatment is discontinued, up to 50% of patients may relapse within the first three months of discontinuation. The mortality rate is increased in bullous pemphigoid due to the elderly population being affected, with a death rate of approximately 10% to 40%.[26]
Media
(Click Image to Enlarge)
References
Vaillant L, Bernard P, Joly P, Prost C, Labeille B, Bedane C, Arbeille B, Thomine E, Bertrand P, Lok C, Roujeau JC. Evaluation of clinical criteria for diagnosis of bullous pemphigoid. French Bullous Study Group. Archives of dermatology. 1998 Sep:134(9):1075-80 [PubMed PMID: 9762017]
Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E, Vaillant L, D'Incan M, Plantin P, Bedane C, Young P, Bernard P, Bullous Diseases French Study Group. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. The New England journal of medicine. 2002 Jan 31:346(5):321-7 [PubMed PMID: 11821508]
Level 1 (high-level) evidenceStavropoulos PG, Soura E, Antoniou C. Drug-induced pemphigoid: a review of the literature. Journal of the European Academy of Dermatology and Venereology : JEADV. 2014 Sep:28(9):1133-40. doi: 10.1111/jdv.12366. Epub 2014 Jan 10 [PubMed PMID: 24404939]
Korman N, Bullous pemphigoid. Journal of the American Academy of Dermatology. 1987 May [PubMed PMID: 3294944]
Level 3 (low-level) evidenceThivolet J, Barthelemy H. Bullous pemphigoid. Seminars in dermatology. 1988 Jun:7(2):91-103 [PubMed PMID: 3153440]
Giudice GJ, Emery DJ, Diaz LA. Cloning and primary structural analysis of the bullous pemphigoid autoantigen BP180. The Journal of investigative dermatology. 1992 Sep:99(3):243-50 [PubMed PMID: 1324962]
Stanley JR, Tanaka T, Mueller S, Klaus-Kovtun V, Roop D. Isolation of complementary DNA for bullous pemphigoid antigen by use of patients' autoantibodies. The Journal of clinical investigation. 1988 Dec:82(6):1864-70 [PubMed PMID: 2461961]
Nishioka K,Hashimoto K,Katayama I,Sarashi C,Kubo T,Sano S, Eosinophilic spongiosis in bullous pemphigoid. Archives of dermatology. 1984 Sep [PubMed PMID: 6383222]
Weigand DA, Clements MK. Direct immunofluorescence in bullous pemphigoid: effects of extent and location of lesions. Journal of the American Academy of Dermatology. 1989 Mar:20(3):437-40 [PubMed PMID: 2645321]
Level 2 (mid-level) evidenceMutasim DF, Adams BB. Immunofluorescence in dermatology. Journal of the American Academy of Dermatology. 2001 Dec:45(6):803-22; quiz 822-4 [PubMed PMID: 11712024]
Lazarova Z, Yancey KB. Reactivity of autoantibodies from patients with defined subepidermal bullous diseases against 1 mol/L salt-split skin. Specificity, sensitivity, and practical considerations. Journal of the American Academy of Dermatology. 1996 Sep:35(3 Pt 1):398-403 [PubMed PMID: 8784276]
Barnadas MA, Gelpi C, Curell R, de Moragas JM, Alomar A. Repeat direct immunofluorescence (DIF) test, using, 1 M NaCl treated skin, in the subepidermal autoimmune bullous diseases that contain IgG at the dermal epidermal junction. Journal of cutaneous pathology. 1999 Jan:26(1):37-41 [PubMed PMID: 10189243]
Alonso-Llamazares J, Rogers RS 3rd, Oursler JR, Calobrisi SD. Bullous pemphigoid presenting as generalized pruritus: observations in six patients. International journal of dermatology. 1998 Jul:37(7):508-14 [PubMed PMID: 9679691]
Level 3 (low-level) evidenceDi Zenzo G, Della Torre R, Zambruno G, Borradori L. Bullous pemphigoid: from the clinic to the bench. Clinics in dermatology. 2012 Jan-Feb:30(1):3-16. doi: 10.1016/j.clindermatol.2011.03.005. Epub [PubMed PMID: 22137222]
Level 3 (low-level) evidenceWaisbourd-Zinman O,Ben-Amitai D,Cohen AD,Feinmesser M,Mimouni D,Adir-Shani A,Zlotkin M,Zvulunov A, Bullous pemphigoid in infancy: Clinical and epidemiologic characteristics. Journal of the American Academy of Dermatology. 2008 Jan [PubMed PMID: 17945382]
Level 3 (low-level) evidencePetronius D, Bergman R. Bullous pemphigoid in two young infants. Pediatric dermatology. 2002 Mar-Apr:19(2):119-21 [PubMed PMID: 11994172]
Level 3 (low-level) evidenceFisler RE, Saeb M, Liang MG, Howard RM, McKee PH. Childhood bullous pemphigoid: a clinicopathologic study and review of the literature. The American Journal of dermatopathology. 2003 Jun:25(3):183-9 [PubMed PMID: 12775979]
Level 3 (low-level) evidenceMartinez-De Pablo MI, González-Enseñat MA, Vicente A, Gilaberte M, Mascaró JM Jr. Childhood bullous pemphigoid: clinical and immunological findings in a series of 4 cases. Archives of dermatology. 2007 Feb:143(2):215-20 [PubMed PMID: 17310001]
Level 3 (low-level) evidenceChiavérini C, Hamel-Teillac D, Gilbert D, Prost Y. Absence of anti-BP180 antibodies in mothers of infants with bullous pemphigoid. The British journal of dermatology. 2006 May:154(5):839-43 [PubMed PMID: 16634883]
Ong E, Goldacre R, Hoang U, Sinclair R, Goldacre M. Associations between bullous pemphigoid and primary malignant cancers: an English national record linkage study, 1999-2011. Archives of dermatological research. 2014 Jan:306(1):75-80. doi: 10.1007/s00403-013-1399-5. Epub 2013 Aug 4 [PubMed PMID: 23912480]
Level 2 (mid-level) evidenceSakuma-Oyama Y, Powell AM, Oyama N, Albert S, Bhogal BS, Black MM. Evaluation of a BP180-NC16a enzyme-linked immunosorbent assay in the initial diagnosis of bullous pemphigoid. The British journal of dermatology. 2004 Jul:151(1):126-31 [PubMed PMID: 15270881]
Level 2 (mid-level) evidenceMariotti F, Grosso F, Terracina M, Ruffelli M, Cordiali-Fei P, Sera F, Zambruno G, Mastrogiacomo A, Di Zenzo G. Development of a novel ELISA system for detection of anti-BP180 IgG and characterization of autoantibody profile in bullous pemphigoid patients. The British journal of dermatology. 2004 Nov:151(5):1004-10 [PubMed PMID: 15541078]
Roujeau JC, Lok C, Bastuji-Garin S, Mhalla S, Enginger V, Bernard P. High risk of death in elderly patients with extensive bullous pemphigoid. Archives of dermatology. 1998 Apr:134(4):465-9 [PubMed PMID: 9554299]
Level 2 (mid-level) evidenceWilliams HC, Wojnarowska F, Kirtschig G, Mason J, Godec TR, Schmidt E, Chalmers JR, Childs M, Walton S, Harman K, Chapman A, Whitham D, Nunn AJ, UK Dermatology Clinical Trials Network BLISTER Study Group. Doxycycline versus prednisolone as an initial treatment strategy for bullous pemphigoid: a pragmatic, non-inferiority, randomised controlled trial. Lancet (London, England). 2017 Apr 22:389(10079):1630-1638. doi: 10.1016/S0140-6736(17)30560-3. Epub 2017 Mar 6 [PubMed PMID: 28279484]
Level 1 (high-level) evidenceBilgiç Temel A, Bassorgun CI, Akman-Karakaş A, Alpsoy E, Uzun S. Successful Treatment of a Bullous Pemphigoid Patient with Rituximab Who Was Refractory to Corticosteroid and Omalizumab Treatments. Case reports in dermatology. 2017 Jan-Apr:9(1):38-44. doi: 10.1159/000452828. Epub 2017 Feb 10 [PubMed PMID: 28413387]
Level 3 (low-level) evidenceFichel F, Barbe C, Joly P, Bedane C, Vabres P, Truchetet F, Aubin F, Michel C, Jegou J, Grange F, Antonicelli F, Bernard P. Clinical and immunologic factors associated with bullous pemphigoid relapse during the first year of treatment: a multicenter, prospective study. JAMA dermatology. 2014 Jan:150(1):25-33. doi: 10.1001/jamadermatol.2013.5757. Epub [PubMed PMID: 24226428]
Level 2 (mid-level) evidencePezzolo E, Naldi L. Epidemiology of major chronic inflammatory immune-related skin diseases in 2019. Expert review of clinical immunology. 2020 Feb:16(2):155-166. doi: 10.1080/1744666X.2020.1719833. Epub 2020 Jan 28 [PubMed PMID: 31962053]