
Introduction
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, is a progressive, paralytic, neurodegenerative disease affecting the upper and lower motor neurons.[1] ALS is the most common motor neuron disease (MND) and has both sporadic and familial forms. Previously, ALS was distinguished from other motor neuron diseases (ie, primary lateral sclerosis, primary muscular atrophy, and progressive bulbar palsy) based on where the patient's first symptoms presented. It is now recognized that ALS presents with diverse clinical heterogeneity.[2] The etiology of ALS is unknown. Numerous possible genetic and sporadic possibilities are suggested. Amyotrophic lateral sclerosis most commonly begins with signs of LMN degeneration affecting the upper extremity but can also present as UMN or bulbar symptoms. Eventually, affected patients will experience respiratory paralysis and, inevitably, death.[1] There is no cure for ALS; however, multiple medications and interventions can help reduce symptoms and improve their quality of life.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
A single and precise etiology governing ALS is lacking. The possible mechanisms described in the literature are genetic mutations, oxidative stress, excitotoxicity, mitochondrial and proteasomal dysfunctions, altered synaptic function, disturbed axonal transport, and neuroinflammation.[3][4] The risk variables include sports with a high incidence of concussions, military jobs, smoking, exposure to heavy metals (lead, manganese), pesticides, neurotoxins (cyanobacteria), and electromagnetic fields.[1][5] It appears that ALS develops as a result of the interaction between both genetic and environmental factors.
Over 120 genes have been implicated in ALS.[1][6] A prevailing thought is that the accumulation of abnormal proteins due to aberrant RNA processing is involved in the development of ALS. Variants of TDP-43 and FUS, two genes for RNA binding proteins, are involved in familial ALS.[7] Patients with variant forms of these genes will have TDP-43 and FUS proteins found primarily in the cytoplasm instead of the nucleus, causing neurodegeneration. In addition, a non-coding stretch of hexanucleotide repeats (to hundreds or thousands) of C9ORF72, which plays a pivotal role in membrane trafficking and autophagy, is the most commonly mutated gene associated with ALS (45% in familial and 7% of sporadic ALS compared to 20% for SOD1).[1][7] Mutations to this gene are also involved in the development of frontotemporal dementia (FTD). Patients with this genetic mutation may develop only ALS, only FTD, or a combination of both. Approximately 50% of people with C9ORF72 expansions develop ALS by age 60 and nearly 100% by age 80.[5]
Superoxide dismutase 1 (SOD1), responsible for converting superoxide radicals to oxygen and hydrogen peroxide, was the first gene discovered in 1993. This prompted the belief that altered protein synthesis is the primary mechanism involved with SOD1 variant ALS (20% of familial cases and 1% of sporadic cases). Cytoskeletal variables such as dynactin 1 (controls retrograde axonal transport), PFN1 (governs the conversion of globular to filamentous actin and nerve extension), and Tubulin 4A (regulates microtubules) are also implicated.[1] The functional deficits at the neuromuscular junction (NMJ) have been observed at least 6 weeks prior to the onset of motor symptoms.[8] SOD1 and profilin 1 (PFN1) are the most commonly applied models for animal studies.[1] The possible role of the brain-gut-microbiota axis in pathogenesis has also been described.[9]
Epidemiology
Of the two forms of amyotrophic lateral sclerosis, 90% to 95% of cases are sporadic (showing a male preponderance of 2:1), with the remainder being familial (male: female ratio of 1:1).[7] The majority of patients with sporadic ALS are found to have an underlying genetic susceptibility (the rest owing to environmental influences).[10]
The lifetime risk of developing ALS is 1:350 for men and 1:400 for women.[5][7] The mean age of involvement is 64.[1][11] This risk is maximum at approximately 75 years.[7] The prevalence of ALS in 22 European countries was 121,028 cases in 2020, with 800,000 cases reported in the US.[1] 380,000 cases worldwide are predicted by 2040.[3] The global incidence is 1 to 2.6 per 100,000 with a prevalence of 4 to 5 per 100,000.[3][5][7] The incidence ranges from 0.26 per 100,000 persons per year in Ecuador to 23.46 per 100,000 per year in Japan. The point prevalence also differs from 1.57 per 100,000 in Iran to 11.80 per 100,000 in the United States.[12]
Pathophysiology
The pathological hallmarks of ALS are the degeneration of the pyramidal Betz cells in the motor cortex, anterior horn cells of the spinal cord (due to retrograde axonal loss), and lower cranial motor nuclei of the brainstem. Gliosis replaces the lost neurons. As a result, the spinal cord atrophies along with the affected muscles. [1][13] A variety of intracellular inclusions are common in degenerating neurons. Bunina bodies are eosinophilic inclusions unique to ALS.[14] The accumulation of TDP-43 is present in most cases of sporadic ALS, FTD, ALS-FTD, and some cases of familial ALS.
The interplay of epigenetics (ie, a modification of gene expression rather than a modification of the genetic code itself), a trigger factor (risk variables described above), a propagating channel (oxidative stress, excitotoxicity, defective RNA processing, intraneuronal protein aggregates, mitochondrial dysfunction, neuroinflammation, or protein misfolding) and final clinical manifestations have also been advocated.[7]
Histopathology
The pathologic hallmarks of the condition were first described by Charcot. There is a loss of motor neurons amid neuroinflammation.[5] Bunina bodies, cytoplasmic transactive response DNA-binding protein 43 (TDP-43) inclusions, and intra-nuclear RNA deposits (C9ORF72 ALS) are also diagnostic.[4]
History and Physical
The clinical presentation of ALS is heterogeneous.[1] The hallmark feature is upper and lower motor neuron signs and symptoms coexist. Upper motor neuron (UMN) findings are hyperreflexia, poor dexterity, incoordination, and spasticity. Dysarthria and dysphagia are common bulbar upper motor neuron findings. Lower motor neuron (LMN) findings are muscle atrophy and fasciculations.[1] Weakness, a hallmark feature, can be attributed to upper or lower motor neurons.
The initial presentation determines the pattern of symptom progression and provides prognostic significance. Most patients present with asymmetric LMN symptoms isolated to the arm or leg.[15] Common initial clinical findings are hand weakness, shoulder girdle weakness, and foot drop. Bulbar onset ALS accounts for 25% of patients. The most common bulbar symptoms are dysarthria and dysphagia. Others are hypernasal speech, laryngospasm, involuntary cheek or tongue biting, and sialorrhea followed by limb involvement in later stages.[1][15] Eye and sphincter muscles are spared until late in the disease.[1] A few patients present with respiratory muscle weakness or generalized weakness in combination with weakness in the bulbar muscles.[5][16]
Many patients (30% to 50%) diagnosed with ALS will develop varying degrees of cognitive impairment, and 15 to 20% of cohorts develop frontotemporal dementia (Pick’s disease).[1] While not presenting with overt dementia, patients can experience changes related to executive function and fluency, as well as behavioral changes such as apathy and disinhibition. Pseudobulbar palsy (ie, inappropriate periods of crying, laughing, or yawning) is also observed due to the involvement of frontopontine motor neurons.[1] Autonomic symptoms (constipation), paresthesias, and pain (due to immobility and muscle cramps) may also occur. In the presence of these symptoms, the diagnosis is ALS-plus syndrome.[17]
As discussed earlier, there is a spectrum of motor neuron diseases (ie, progressive muscular atrophy and primary lateral sclerosis). The involvement of upper and lower motor neurons currently defines each. Although debated, some experts believe progressive muscular atrophy (PMA) represents a form of ALS. The disease is progressive and initially exclusively involves the LMN. Many patients progress and develop clinical signs and symptoms of UMN disease, at which point the diagnosis becomes lower motor neuron-onset ALS. Interestingly, in patients who never show clinical evidence of UMN involvement, corticospinal tract involvement is detected at autopsy in more than 50% of patients with an antemortem diagnosis of PMA.[18] Primary lateral sclerosis (PLS) initially presents with exclusively UMN symptoms. Most will eventually develop LMN symptoms, and the diagnosis becomes upper motor neuron-dominant ALS.[19] These patients have a slower progression of symptoms, lack weight loss, and LMN signs and symptoms in the first four years of the disease.
The clinical phenotypes of traditional ALS are typical spinal onset, bulbar onset, flail arm syndrome, flail leg phenotype, hemiplegic, pseudo-polyneuritic phenotype, mixed, and thoracic/respiratory onset variants.[5][20] Flail arm syndrome ( or brachial amyotrophic diplegia), is characterized by LMN weakness and wasting. It usually causes symmetric proximal weakness in the upper limbs.[21] Another variant is flail leg syndrome (pseudo-polyneuritic variant), also characterized by weakness and wasting, but of the lower extremities and with distal onset. Patients with flail arm and leg syndrome have a slower rate of progression to the involvement of other body segments and respiratory muscle weakness.[22] These variants are difficult to differentiate from typical ALS as the diagnostic criteria are primarily the length of time symptoms remain localized to the initial extremity regions.
Progression of ALS is usually linear, without episodes of remissions or exacerbations. While the rate of progression varies between individuals, the pattern of progression is relatively predictable. The most common pattern in patients is unilateral limb onset (again, the predominant form), which progresses to include the contralateral limb, then the other ipsilateral extremity (ie, the leg if the initial weakness was in the arm), followed by the other contralateral extremity, before ultimately affecting the bulbar muscles.
Evaluation
Due to its variable presentation, lack of diagnostic markers, and similarity to various motor neuron diseases, ALS is primarily a clinical diagnosis. The revised El Escorial criteria were developed to properly place patients in clinical trials and provide diagnostic certainty. The revised El Escorial criteria were subsequently altered to incorporate relevant electrophysiological changes (equivalent to clinical LMN findings) and help improve diagnostic sensitivity.[5][23] Specifically, the Awaji criteria state that diagnosing ALS requires clinical, electrophysical, or neuropathological evidence of combined lower and upper motor neuron degeneration with progression and excludes treatable clinical mimics.[24]
All patients with motor neuron disease should undergo nerve conduction studies and electromyography (EMG). They are helpful in further supporting the diagnosis if the clinical picture is unclear. EMG contributes to the diagnosis in up to 90% of cases.[16]. Expected electromyography (EMG) findings are acute denervation (fibrillation and positive sharp waves), chronic denervation (long-duration, complex motor unit action potentials [MUAP]), and chronic reinnervation (large amplitude MUAP).[1][25] Sensory action potentials will be normal. Motor nerve action potential amplitudes may be low before weakness is clinically evident.[26] EMG criteria for diagnosing ALS are acute or chronic denervation in at least three spinal levels (bulbar, cervical, thoracic, or lumbosacral). If three spinal levels are not abnormal, acute or chronic denervation must be evident in three extremities involving at least two muscles supplied by two different nerve roots in each extremity. There should be no features of demyelination (prolonged distal latencies >30%, conduction block, or reduced velocity <70%).[5]
The role of radiology in evaluating ALS is primarily to exclude other potential causes of the patient's symptoms. MRI is the imaging study of choice. MRI will typically be normal with ALS. Occasionally, decreased signal intensity within the motor cortex on T2-weighted images (T2WI) and well-defined lesions of increased signal intensity can be visible within the corticospinal tracts on T2WI. The "motor band sign" is a decreased signal visible on susceptibility-weighted imaging across the precentral gyrus caused by the accumulation of iron within the precentral gyrus.[27][28] Findings indicative of upper motor neuronal disease have been noted utilizing experimental advanced MRI techniques such as spectroscopy and diffusion tensor imaging (DTI).[28][29] Fiber density and cross-section provided the greatest sensitivity to longitudinal change.[30] MR spectroscopy with a reduced N-acetyl aspartate-inositol ratio is very specific.[3][31][32] T2/FLAIR hyperintensities along the corticospinal tract and internal capsule are also affirmative.
To monitor disease progression, the King's staging system was developed.
The grading of the King's system includes:
- Stage 1-Symptom onset with involvement of the first region
- Stage 2A-Diagnosis
- Stage 2B-Involvement of the second region
- Stage 3-Involvement of the third region
- Stage 4A-Need for gastrostomy
- Stage 4B-Need for noninvasive ventilation
- Stage 5-Death[5][20]
The majority of affected patients are diagnosed within King's Stage 2, with only 16% initially assessed by a neurologist.[16] Healthcare providers can use the 'thinkALS' and 'MND Red Flag' tools to help with earlier diagnosis.[16] PRECISION-ALS (a large European research initiative) was formed to ensure precision medicine.[33]
Due to emerging genotype-specific therapies in clinical trials, genetic testing is encouraged upon diagnosis, particularly for the SOD1 and C9ORF72 genotypes. Genetic testing can be used as a screening tool for patients with a family history of autosomal dominant disease and prognostic implications. This can also help to elucidate the degree of penetrance. ALS polygenic scores can account for cumulative genetic risk in populations and reflect disease-relevant pathways.[34]
Treatment / Management
The treatment of ALS is multidisciplinary and involves physical, occupational, and speech therapists, home ventilation therapy, nutrition support, psychotherapy, training for caregivers, cognitive screening, and end-of-life care.[3][20] Symptomatic management is the mainstay of treatment.[35](A1)
Riluzole, Edaravone, and sodium phenylbutyrate are the only approved medications to treat ALS but provide limited survival benefits.[1][3] Riluzole, 50mg bid, is recommended for all patients with ALS and is thought to reduce glutamate-induced excitotoxicity. Riluzole is the only medication that improves overall survival (74% vs. 58% in the placebo group at 12 months) and slows the progression of symptoms (ie, survival without tracheostomy (57% vs. 50% at 18 months). Riluzole is most effective in patients who have been symptomatic for less than 5 years, have a vital capacity (VC) >60% of the predicted value, and do not require a tracheostomy.[36] It is important to note that the elimination of riluzole will be affected by CYP1A2 inhibitors like caffeine and theophylline (decrease the rate of elimination of riluzole). Transaminase elevation occurs in approximately 50% of patients. Monitor transaminases monthly for the first three months and every three months after. (B2)
Edaravone is a free radical scavenger that is thought to reduce oxidative stress and slow functional progression. Edaravone provides the most benefit in patients with early ALS (ie, less than two years of symptoms, living independently, VC >80%, and scores of 2 or more in all items of ALSFRS-R) with probable or definite ALS (revised El Escorial criteria). One study showed approximately 33% slower functional decline in patients at 24 weeks follow-up.[37] Edaravone treatment is expensive ($146,000 annually). Edaravone should be used with caution in patients with asthma. It contains sodium bisulfite, which may cause asthmatic reactions in up to 5% of patients. (A1)
Sodium phenylbutyrate is a histone deacetylase inhibitor that decreases the stress response in the endoplasmic reticulum. Combined with taurursodiol (which increases the threshold for cellular apoptosis), it decreases neuronal destruction. The result is a slower decline in function. Sodium phenylbutyrate-taurursodiol is administered intrathecally and may result in myelitis, radiculitis, aseptic meningitis, and elevated intracranial pressure.
Patients with ALS will suffer from chronic respiratory failure due to weakness of the diaphragmatic and intercostal muscles. Pulmonary function tests are done every three months. As this weakness is progressive, an early, careful discussion regarding respiratory management and future options such as tracheostomy, chronic ventilatory support, and noninvasive ventilation (NIV) is warranted. The discussion should evolve with the progression of symptoms and patient readiness. NIV will be helpful when the vital capacity (VC) is less than 50%, sniff nasal inspiratory pressure (SNIP) < 40 cm H2O, pCO2 > 45mmHg, significant desaturations are noted on overnight oximetry, or when evidence of respiratory weakness is present.[20] The use of NIV prolongs survival.[5] Invasive ventilation is considered when NIV is no longer tolerated, or when the patient continues to experience hypoxia or hypercarbia despite NIV. Survival after beginning invasive ventilation is approximately 30 months. Patients should understand that they can discontinue therapy at any time during the treatment process. Immunization with the annual influenza and pneumococcal vaccines is important. (A1)
Initially managed with diet modifications, progressive weakening of the muscles of mastication and swallowing results in dysphagia. Weight loss (particularly within the first two years after diagnosis) is associated with a poorer prognosis. Emphasis should be placed on the intake of calorically dense foods and supplemental nutritional beverages.[38] Enteral tube feeding is indicated when weight loss exceeds 10%, BMI is less than 18.5 kg/m2, swallowing impairments are observed during instrumental tests, or the VC is <50%.[20] PEG is superior in terms of quality-of-life measures. If the patient elects to have a G-tube placed, this should be completed before the VC becomes <50% of the predicted value to avoid increased morbidity associated with the procedure itself.[36] Tertiary ALS centers increase six-month survival as well as reduce time to noninvasive ventilation (NIV) and Gastrostomy.[3] (A1)
As the disease progresses, there will be increased dysarthria and the inability to communicate. Speech therapy provides limited benefit, and appropriate alternative communication methods (writing, alphabet boards, and electronic assistive communication devices) are needed.[39](B2)
Frequent and painful muscle spasms are common. Mexiletine (a sodium channel-blocking antiarrhythmic drug) is effective at a 150 mg BID dose.[40][41] Other options include levetiracetam, gabapentin, baclofen, and tizanidine.[42] When oral therapy is impractical, botulinum toxin injections into the spastic muscles can be helpful. As weakness and functional decline inevitably progress, patients use assistive devices (canes, orthoses, crutches, and eventually wheelchairs), removable headrests in those with neck weakness, specialized utensils and holders, and eventually a pressure-relieving mattress with frequent repositioning to prevent pressure ulcers.[43] Sialorrhea is very common and can be treated with atropine, hyoscyamine, amitriptyline, glycopyrrolate, botulinum toxin injections into salivary glands, and even low-dose radiation therapy in those with refractory symptoms.[35][44] (A1)
AVP-923 (dextromethorphan-quinidine 20mg/10mg) has proven effective in patients with pseudobulbar affect.[45] Less rigorous evidence has supported the use of amitriptyline and fluvoxamine in smaller trials. Pain is treated based on its underlying cause. Non-steroidal anti-inflammatory medications, paracetamol, intra-articular steroids, or lidocaine injections are recommended for nociceptive pain, whereas gabapentin, pregabalin, tricyclic antidepressants, and morphine are advised for neuropathic pain. Mexiletine, levetiracetam, and quinine sulfate help patients with cramps. Assistive devices such as special mattresses, pillows, and wheelchairs may help to prevent pain. Ultimately, many patients require nonopioid analgesics and anti-inflammatory medications, and when these fail, opioids become the mainstay of pain treatment.[46] (A1)
Depression is common, and treatment improves the quality of life. Amitriptyline is commonly used as it can also treat other symptoms such as insomnia, sialorrhea, and pseudobulbar affect. Selective serotonin reuptake inhibitors are another alternative if the TCAs are not tolerated. The issue of end-of-life care and advanced directives should be broached soon after diagnosis. Treating dyspnea and anxiety is essential. In addition to NIV, relaxation techniques, psychosocial support, morphine, and benzodiazepines are helpful. Insomnia should be first addressed by treating the underlying cause (ie, dyspnea, pain, or anxiety). Tricyclic antidepressants and benzodiazepines are medication options.[20] Hospice and palliative care providers can provide many resources that are not available or as readily available in the home and can increase the likelihood of a peaceful death.[47] Ongoing trials focus on nanoparticles' role, viral vectors, monoclonal-antibody, and stem cells.[1][3] Artificial intelligence (AI) and machine learning may prove to be a prodigy in management.[48](A1)
Differential Diagnosis
The LMN Mimics
- Benign fasciculations
- Inclusion body myopathy
- Multifocal motor neuropathy
- Monomelic amyotrophy
- Neuralgic myopathy
- Hirayama disease
- Spinobobulbar muscular atrophy
- Distal spinal atrophy syndrome (Charcot-Marie-Tooth disease)
The UMN Mimics
- Hereditary spastic paraplegia
- Adrenomyeloneuropathy
- Late-onset Tay-Sachs disease
- Polyglucosan body disease
- HIV myelopathy
- Multiple sclerosis
- Myasthenia gravis
- Nutritional myeloneuropathies
- Post-polio syndrome
A Mixed Pattern
Prognosis
There is no cure for ALS.[5][7] The median survival is 3 to 5 years. Approximately 30% of patients survive 5 years and 10% ten years. Riluzole provides a survival benefit averaging 3 months, with edaravone showing a 33% reduction in the decline of the ALS Functional Rating Scale–Revised (ALSFRS-R) at 6 months.
Factors governing improved survival include mild obesity at the time of diagnosis, younger age at onset, higher ALS Functional Rating Scale score, better forced vital capacity (FVC), and limb (rather than bulbar) symptoms on presentation.[53][54] Patients with the flail-arm syndrome phenotype also survive longer.[1] SOD1 mutations show predominantly lower limb onset, and TARDP mutations have upper limb onset. Lower expression of EPHA4 connotes a more prolonged survival. Increased age at onset, a short delay from symptom onset to diagnosis, and the earlier use of NIV are all prognostic indicators of a more aggressive progression.[2] Accelerated progression is also observed in the A4V mutation of SOD1 and P525L mutation of FUS/TLS, showing a fulminant childhood pattern.[1] FUS mutations have an earlier age of onset and shorter lifespan.[7]
Complications
There are numerous complications associated with amyotrophic lateral sclerosis, including respiratory decline with eventual need for ventilatory support, dysphagia, dysarthria, malnutrition, muscle spasms, spasticity, fatigue, functional decline due to muscular weakness, sialorrhea, thick mucus secretions, and pseudobulbar affect. Aspiration pneumonia and respiratory insufficiency are the most common causes of death.[7] In addition, some complications may arise due to the medications meant to manage disease symptoms. Mexiletine can cause gastrointestinal upset and arrhythmias. Riluzole can lead to transaminitis and asthenia, whereas edaravone can cause gait disturbance and headache.
Postoperative and Rehabilitation Care
Early intervention is crucial to maximize functional mobility and longevity. Comprehensive initial assessments of balance, gait, muscle strength and tone, activity tolerance, and performance of activities of daily living (ADLs) are of paramount significance. Fall prevention strategies (including assistive devices and environmental modifications) and energy conservation techniques are required to combat weakness and fatigue. Orthoses and adaptive equipment may also be utilized to further independence with ADLs.
Collaboration among healthcare providers, therapists, and caregivers is vital for comprehensive disease management. Throughout the course of care, it is important not only to be mindful of the natural history of the disease but also to focus on maximizing the patient’s comfort, functionality, and quality of life.[55][56] Patients' and caregivers' education and access to adequate social support is equally critical.[20]
Deterrence and Patient Education
ALS is a progressive, degenerative disease that causes muscle weakness, eventually affecting most muscles in the body. Some people inherit ALS from their family, but most develop it for various other reasons. Many people start with symptoms in just one arm or one leg. Others begin with difficulty speaking, swallowing, or breathing. Eventually, all patients will develop weakness in the muscles that help them swallow and breathe, requiring assistance with nutrition and breathing. The average patient survives 3 to 5 years after diagnosis, but longer survival times are possible. Speech therapists, occupational therapists, physical therapists, neurologists, nurses, respiratory therapists, dieticians, palliative care providers, and social workers are all involved in the care of a patient with ALS. Receiving the diagnosis of ALS can be overwhelming, and patients who seek care at multidisciplinary ALS clinics have improved survival over those followed by a single healthcare provider. While there is no cure, multiple medications, and management strategies exist to prolong survival and alleviate symptoms.
Pearls and Other Issues
One classic presentation of ALS may not exist. Personalized therapy needs to be implemented.[3] Each patient must be evaluated for ALS mimics. Treatment at multidisciplinary ALS clinics improve patient outcomes.
Enhancing Healthcare Team Outcomes
ALS causes significant debilitation and eventually death. The clinical presentation of ALS is broad, and each patient's care plan should be individualized through a multidisciplinary approach. Treatment teams involve physicians, occupational, physical, speech, and respiratory therapists, dieticians, nurses, social workers, and case managers. Each member has a unique opportunity to recognize changes in the patient's condition and can notify other providers, allowing for quicker interventions.
The neurologist frames the treatment plan while the physiatrist promotes patients' physical capabilities. Physical and occupational therapists build muscle endurance and assist in maintaining routine physical activities. The pulmonologist and respiratory therapist facilitate respiratory independence. A dietitian and gastroenterologist assist in nutritional support and weight management. A speech therapist helps with swallowing and communication, and a neuropsychologist boosts mental health support for the patient and their care providers. A social worker will address the logistical issues about finances, insurance, transportation, employment, home care, and community resources and programs. When patients with ALS are cared for by such an interprofessional team, there have been significant, demonstrable benefits in both the quality of life and the length of survival.[57][58] Such an approach has been shown to improve one-year mortality by almost 30% and overall lifespan by almost 8 months.[58] The addition of telehealth and ALS home call programs may help provide tailored care to the health needs of patients with ALS.[59] Genetic testing and gene-specific treatment may be pivotal in the future to unlock the disease process and its management.[3]
Media
(Click Image to Enlarge)
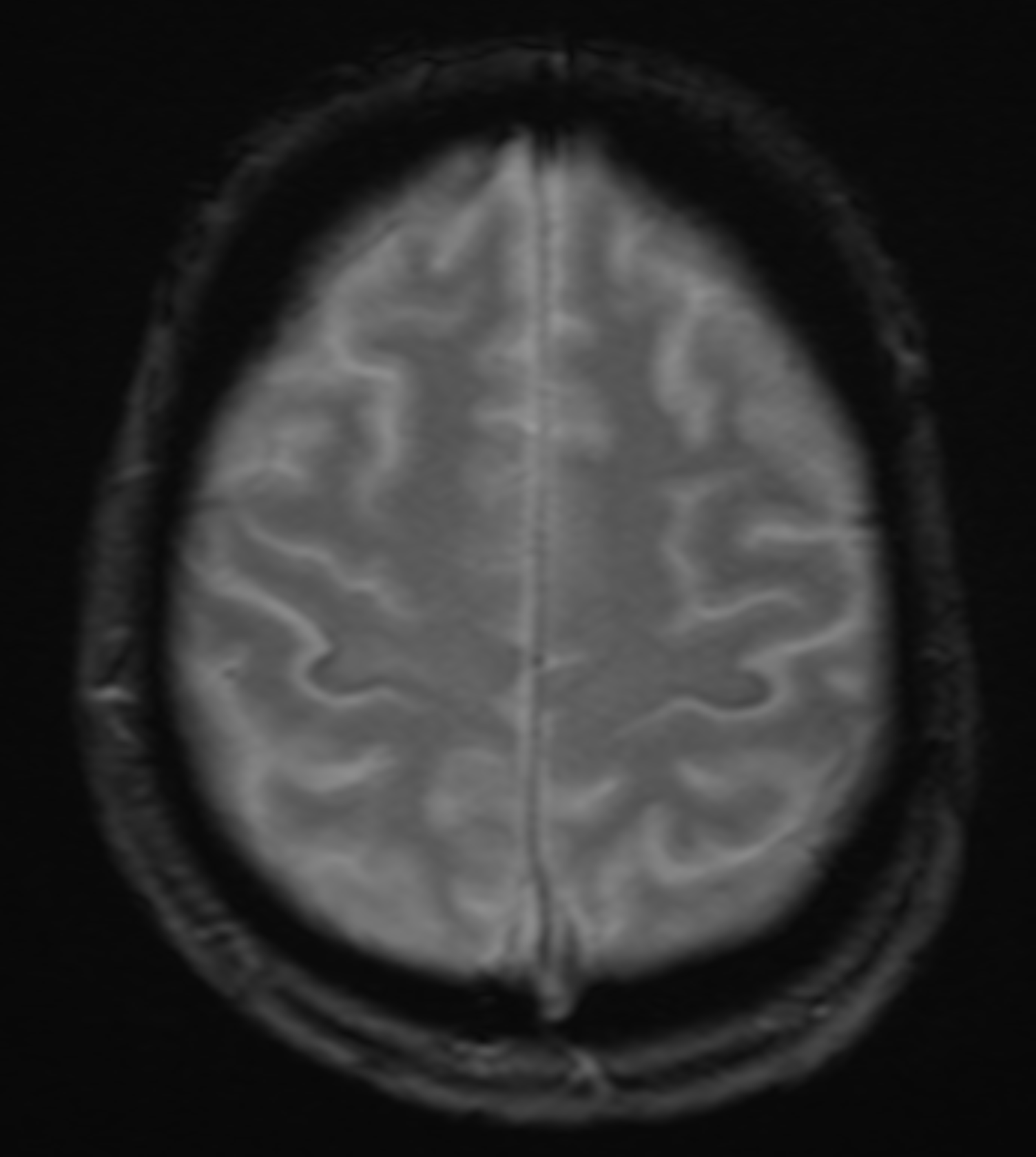
MRI GRE sequence demonstrating iron deposition in the precentral gyrus (motor band sign) in a patient with amyotrophic lateral sclerosis.
Contributed by Ryan Brotman, DO
References
Brown RH, Al-Chalabi A. Amyotrophic Lateral Sclerosis. The New England journal of medicine. 2017 Jul 13:377(2):162-172. doi: 10.1056/NEJMra1603471. Epub [PubMed PMID: 28700839]
Alves I, Gromicho M, Oliveira Santos M, Pinto S, Pronto-Laborinho A, Swash M, de Carvalho M. Demographic changes in a large motor neuron disease cohort in Portugal: a 27 year experience. Amyotrophic lateral sclerosis & frontotemporal degeneration. 2023 Jun 9:():1-11. doi: 10.1080/21678421.2023.2220747. Epub 2023 Jun 9 [PubMed PMID: 37295966]
Tzeplaeff L, Wilfling S, Requardt MV, Herdick M. Current State and Future Directions in the Therapy of ALS. Cells. 2023 May 31:12(11):. doi: 10.3390/cells12111523. Epub 2023 May 31 [PubMed PMID: 37296644]
Level 3 (low-level) evidenceMargeta M. Neuromuscular disease: 2023 update. Free neuropathology. 2023 Jan:4():. pii: 4-2. doi: 10.17879/freeneuropathology-2023-4682. Epub 2023 Feb 27 [PubMed PMID: 37283936]
Quinn C, Elman L. Amyotrophic Lateral Sclerosis and Other Motor Neuron Diseases. Continuum (Minneapolis, Minn.). 2020 Oct:26(5):1323-1347. doi: 10.1212/CON.0000000000000911. Epub [PubMed PMID: 33003004]
Wang H, Guan L, Deng M. Recent progress of the genetics of amyotrophic lateral sclerosis and challenges of gene therapy. Frontiers in neuroscience. 2023:17():1170996. doi: 10.3389/fnins.2023.1170996. Epub 2023 May 12 [PubMed PMID: 37250416]
Morgan S, Orrell RW. Pathogenesis of amyotrophic lateral sclerosis. British medical bulletin. 2016 Sep:119(1):87-98. doi: 10.1093/bmb/ldw026. Epub 2016 Jul 22 [PubMed PMID: 27450455]
McIntosh J, Mekrouda I, Dashti M, Giuraniuc CV, Banks RW, Miles GB, Bewick GS. Development of abnormalities at the neuromuscular junction in the SOD1-G93A mouse model of ALS: dysfunction then disruption of postsynaptic structure precede overt motor symptoms. Frontiers in molecular neuroscience. 2023:16():1169075. doi: 10.3389/fnmol.2023.1169075. Epub 2023 May 19 [PubMed PMID: 37273905]
Chen S, Cai X, Lao L, Wang Y, Su H, Sun H. Brain-Gut-Microbiota Axis in Amyotrophic Lateral Sclerosis: A Historical Overview and Future Directions. Aging and disease. 2023 Jun 6:():. doi: 10.14336/AD.2023.0524. Epub 2023 Jun 6 [PubMed PMID: 37307822]
Level 3 (low-level) evidenceYu B, Pamphlett R. Environmental insults: critical triggers for amyotrophic lateral sclerosis. Translational neurodegeneration. 2017:6():15. doi: 10.1186/s40035-017-0087-3. Epub 2017 Jun 16 [PubMed PMID: 28638596]
Worms PM. The epidemiology of motor neuron diseases: a review of recent studies. Journal of the neurological sciences. 2001 Oct 15:191(1-2):3-9 [PubMed PMID: 11676986]
Wolfson C, Gauvin DE, Ishola F, Oskoui M. Global Prevalence and Incidence of Amyotrophic Lateral Sclerosis: A Systematic Review. Neurology. 2023 Aug 8:101(6):e613-e623. doi: 10.1212/WNL.0000000000207474. Epub 2023 Jun 12 [PubMed PMID: 37308302]
Level 1 (high-level) evidenceSaberi S, Stauffer JE, Schulte DJ, Ravits J. Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurologic clinics. 2015 Nov:33(4):855-76. doi: 10.1016/j.ncl.2015.07.012. Epub [PubMed PMID: 26515626]
Piao YS, Wakabayashi K, Kakita A, Yamada M, Hayashi S, Morita T, Ikuta F, Oyanagi K, Takahashi H. Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain pathology (Zurich, Switzerland). 2003 Jan:13(1):10-22 [PubMed PMID: 12580541]
Level 3 (low-level) evidenceZarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A. A comprehensive review of amyotrophic lateral sclerosis. Surgical neurology international. 2015:6():171. doi: 10.4103/2152-7806.169561. Epub 2015 Nov 16 [PubMed PMID: 26629397]
Gwathmey KG, Corcia P, McDermott CJ, Genge A, Sennfält S, de Carvalho M, Ingre C. Diagnostic delay in amyotrophic lateral sclerosis. European journal of neurology. 2023 Sep:30(9):2595-2601. doi: 10.1111/ene.15874. Epub 2023 May 30 [PubMed PMID: 37209406]
McCluskey LF, Elman LB, Martinez-Lage M, Van Deerlin V, Yuan W, Clay D, Siderowf A, Trojanowski JQ. Amyotrophic lateral sclerosis-plus syndrome with TAR DNA-binding protein-43 pathology. Archives of neurology. 2009 Jan:66(1):121-4. doi: 10.1001/archneur.66.1.121. Epub [PubMed PMID: 19139310]
Level 3 (low-level) evidenceRowland LP. Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle & nerve. 2010 Feb:41(2):161-5. doi: 10.1002/mus.21565. Epub [PubMed PMID: 20082312]
Gordon PH, Cheng B, Katz IB, Pinto M, Hays AP, Mitsumoto H, Rowland LP. The natural history of primary lateral sclerosis. Neurology. 2006 Mar 14:66(5):647-53 [PubMed PMID: 16534101]
Level 2 (mid-level) evidenceBoostani R, Olfati N, Shamshiri H, Salimi Z, Fatehi F, Hedjazi SA, Fakharian A, Ghasemi M, Okhovat AA, Basiri K, Haghi Ashtiani B, Ansari B, Raissi GR, Khatoonabadi SA, Sarraf P, Movahed S, Panahi A, Ziaadini B, Yazdchi M, Bakhtiyari J, Nafissi S. Iranian clinical practice guideline for amyotrophic lateral sclerosis. Frontiers in neurology. 2023:14():1154579. doi: 10.3389/fneur.2023.1154579. Epub 2023 Jun 2 [PubMed PMID: 37333000]
Level 1 (high-level) evidenceCouratier P, Truong C, Khalil M, Devière F, Vallat JM. Clinical features of flail arm syndrome. Muscle & nerve. 2000 Apr:23(4):646-8 [PubMed PMID: 10716778]
Level 3 (low-level) evidenceWijesekera LC, Mathers S, Talman P, Galtrey C, Parkinson MH, Ganesalingam J, Willey E, Ampong MA, Ellis CM, Shaw CE, Al-Chalabi A, Leigh PN. Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology. 2009 Mar 24:72(12):1087-94. doi: 10.1212/01.wnl.0000345041.83406.a2. Epub [PubMed PMID: 19307543]
Level 2 (mid-level) evidenceCosta J, Swash M, de Carvalho M. Awaji criteria for the diagnosis of amyotrophic lateral sclerosis:a systematic review. Archives of neurology. 2012 Nov:69(11):1410-6 [PubMed PMID: 22892641]
Level 1 (high-level) evidenceBrooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis and other motor neuron disorders : official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2000 Dec:1(5):293-9 [PubMed PMID: 11464847]
Level 1 (high-level) evidenceKrivickas LS. Amyotrophic lateral sclerosis and other motor neuron diseases. Physical medicine and rehabilitation clinics of North America. 2003 May:14(2):327-45 [PubMed PMID: 12795519]
Gooch CL, Shefner JM. ALS surrogate markers. MUNE. Amyotrophic lateral sclerosis and other motor neuron disorders : official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2004 Sep:5 Suppl 1():104-7 [PubMed PMID: 15512887]
Level 2 (mid-level) evidenceKwan JY, Jeong SY, Van Gelderen P, Deng HX, Quezado MM, Danielian LE, Butman JA, Chen L, Bayat E, Russell J, Siddique T, Duyn JH, Rouault TA, Floeter MK. Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 tesla MRI and pathology. PloS one. 2012:7(4):e35241. doi: 10.1371/journal.pone.0035241. Epub 2012 Apr 17 [PubMed PMID: 22529995]
Wang S, Melhem ER, Poptani H, Woo JH. Neuroimaging in amyotrophic lateral sclerosis. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2011 Jan:8(1):63-71. doi: 10.1007/s13311-010-0011-3. Epub [PubMed PMID: 21274686]
Anand T, Ishaque A, Ta D, Khan MU, Bharti K, Wu A, Krebs D, Beaulieu C, Seres P, Kalra S. Characterization of white matter alterations using diffusion kurtosis imaging in patients with amyotrophic lateral sclerosis. Brain and behavior. 2023 Jul:13(7):e3102. doi: 10.1002/brb3.3102. Epub 2023 Jun 6 [PubMed PMID: 37279166]
Pisharady PK, Eberly LE, Adanyeguh IM, Manousakis G, Guliani G, Walk D, Lenglet C. Multimodal MRI improves diagnostic accuracy and sensitivity to longitudinal change in amyotrophic lateral sclerosis. Communications medicine. 2023 Jun 16:3(1):84. doi: 10.1038/s43856-023-00318-5. Epub 2023 Jun 16 [PubMed PMID: 37328685]
Verma G, Woo JH, Chawla S, Wang S, Sheriff S, Elman LB, McCluskey LF, Grossman M, Melhem ER, Maudsley AA, Poptani H. Whole-brain analysis of amyotrophic lateral sclerosis by using echo-planar spectroscopic imaging. Radiology. 2013 Jun:267(3):851-7. doi: 10.1148/radiol.13121148. Epub 2013 Jan 29 [PubMed PMID: 23360740]
Level 2 (mid-level) evidenceGovind V, Sharma KR, Maudsley AA, Arheart KL, Saigal G, Sheriff S. Comprehensive evaluation of corticospinal tract metabolites in amyotrophic lateral sclerosis using whole-brain 1H MR spectroscopy. PloS one. 2012:7(4):e35607. doi: 10.1371/journal.pone.0035607. Epub 2012 Apr 23 [PubMed PMID: 22539984]
McFarlane R, Galvin M, Heverin M, Mac Domhnaill É, Murray D, Meldrum D, Bede P, Bolger A, Hederman L, Impey S, Stephens G, O'Meara C, Wade V, Al-Chalabi A, Chiò A, Corcia P, van Damme P, Ingre C, McDermott C, Povedanos M, van den Berg L, Hardiman O. PRECISION ALS-an integrated pan European patient data platform for ALS. Amyotrophic lateral sclerosis & frontotemporal degeneration. 2023 Aug:24(5-6):389-393. doi: 10.1080/21678421.2023.2215838. Epub 2023 May 23 [PubMed PMID: 37221648]
Dou J, Bakulski K, Guo K, Hur J, Zhao L, Saez-Atienzar S, Stark A, Chia R, García-Redondo A, Rojas-Garcia R, Vázquez Costa JF, Fernandez Santiago R, Bandres-Ciga S, Gómez-Garre P, Periñán MT, Mir P, Pérez-Tur J, Cardona F, Menendez-Gonzalez M, Riancho J, Borrego-Hernández D, Galán-Dávila L, Infante Ceberio J, Pastor P, Paradas C, Dols-Icardo O, Traynor BJ, Feldman EL, Goutman SA, Spanish Neurological Consortium. Cumulative Genetic Score and C9orf72 Repeat Status Independently Contribute to Amyotrophic Lateral Sclerosis Risk in 2 Case-Control Studies. Neurology. Genetics. 2023 Aug:9(4):e200079. doi: 10.1212/NXG.0000000000200079. Epub 2023 May 31 [PubMed PMID: 37293291]
Level 2 (mid-level) evidenceMiller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, Kalra S, Katz JS, Mitsumoto H, Rosenfeld J, Shoesmith C, Strong MJ, Woolley SC, Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009 Oct 13:73(15):1227-33. doi: 10.1212/WNL.0b013e3181bc01a4. Epub [PubMed PMID: 19822873]
Level 2 (mid-level) evidenceMiller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, Kalra S, Katz JS, Mitsumoto H, Rosenfeld J, Shoesmith C, Strong MJ, Woolley SC, Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009 Oct 13:73(15):1218-26. doi: 10.1212/WNL.0b013e3181bc0141. Epub [PubMed PMID: 19822872]
Level 2 (mid-level) evidenceWriting Group, Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. The Lancet. Neurology. 2017 Jul:16(7):505-512. doi: 10.1016/S1474-4422(17)30115-1. Epub 2017 May 15 [PubMed PMID: 28522181]
Level 1 (high-level) evidenceKasarskis EJ, Mendiondo MS, Matthews DE, Mitsumoto H, Tandan R, Simmons Z, Bromberg MB, Kryscio RJ, ALS Nutrition/NIPPV Study Group. Estimating daily energy expenditure in individuals with amyotrophic lateral sclerosis. The American journal of clinical nutrition. 2014 Apr:99(4):792-803. doi: 10.3945/ajcn.113.069997. Epub 2014 Feb 12 [PubMed PMID: 24522445]
Körner S, Sieniawski M, Kollewe K, Rath KJ, Krampfl K, Zapf A, Dengler R, Petri S. Speech therapy and communication device: impact on quality of life and mood in patients with amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis & frontotemporal degeneration. 2013 Jan:14(1):20-5. doi: 10.3109/17482968.2012.692382. Epub 2012 Aug 7 [PubMed PMID: 22871079]
Level 2 (mid-level) evidenceWeiss MD, Macklin EA, Simmons Z, Knox AS, Greenblatt DJ, Atassi N, Graves M, Parziale N, Salameh JS, Quinn C, Brown RH Jr, Distad JB, Trivedi J, Shefner JM, Barohn RJ, Pestronk A, Swenson A, Cudkowicz ME, Mexiletine ALS Study Group. A randomized trial of mexiletine in ALS: Safety and effects on muscle cramps and progression. Neurology. 2016 Apr 19:86(16):1474-81. doi: 10.1212/WNL.0000000000002507. Epub 2016 Feb 24 [PubMed PMID: 26911633]
Level 1 (high-level) evidenceOskarsson B, Moore D, Mozaffar T, Ravits J, Wiedau-Pazos M, Parziale N, Joyce NC, Mandeville R, Goyal N, Cudkowicz ME, Weiss M, Miller RG, McDonald CM. Mexiletine for muscle cramps in amyotrophic lateral sclerosis: A randomized, double-blind crossover trial. Muscle & nerve. 2018 Mar 6:():. doi: 10.1002/mus.26117. Epub 2018 Mar 6 [PubMed PMID: 29510461]
Level 1 (high-level) evidenceBedlack RS, Pastula DM, Hawes J, Heydt D. Open-label pilot trial of levetiracetam for cramps and spasticity in patients with motor neuron disease. Amyotrophic lateral sclerosis : official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2009 Aug:10(4):210-5. doi: 10.1080/17482960802430773. Epub [PubMed PMID: 18821142]
Level 3 (low-level) evidenceBorasio GD, Voltz R, Miller RG. Palliative care in amyotrophic lateral sclerosis. Neurologic clinics. 2001 Nov:19(4):829-47 [PubMed PMID: 11854102]
Stone CA, O'Leary N. Systematic review of the effectiveness of botulinum toxin or radiotherapy for sialorrhea in patients with amyotrophic lateral sclerosis. Journal of pain and symptom management. 2009 Feb:37(2):246-58. doi: 10.1016/j.jpainsymman.2008.02.006. Epub 2008 Aug 3 [PubMed PMID: 18676117]
Level 1 (high-level) evidenceBrooks BR, Thisted RA, Appel SH, Bradley WG, Olney RK, Berg JE, Pope LE, Smith RA, AVP-923 ALS Study Group. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial. Neurology. 2004 Oct 26:63(8):1364-70 [PubMed PMID: 15505150]
Level 1 (high-level) evidenceChiò A, Mora G, Lauria G. Pain in amyotrophic lateral sclerosis. The Lancet. Neurology. 2017 Feb:16(2):144-157. doi: 10.1016/S1474-4422(16)30358-1. Epub 2016 Dec 8 [PubMed PMID: 27964824]
Ganzini L, Johnston WS, Silveira MJ. The final month of life in patients with ALS. Neurology. 2002 Aug 13:59(3):428-31 [PubMed PMID: 12177378]
Tavazzi E, Longato E, Vettoretti M, Aidos H, Trescato I, Roversi C, Martins AS, Castanho EN, Branco R, Soares DF, Guazzo A, Birolo G, Pala D, Bosoni P, Chiò A, Manera U, de Carvalho M, Miranda B, Gromicho M, Alves I, Bellazzi R, Dagliati A, Fariselli P, Madeira SC, Di Camillo B. Artificial intelligence and statistical methods for stratification and prediction of progression in amyotrophic lateral sclerosis: A systematic review. Artificial intelligence in medicine. 2023 Aug:142():102588. doi: 10.1016/j.artmed.2023.102588. Epub 2023 May 20 [PubMed PMID: 37316101]
Level 1 (high-level) evidenceHardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, van den Berg LH. Amyotrophic lateral sclerosis. Nature reviews. Disease primers. 2017 Oct 5:3():17071. doi: 10.1038/nrdp.2017.71. Epub 2017 Oct 5 [PubMed PMID: 28980624]
Pradhan S. Bilaterally symmetric form of Hirayama disease. Neurology. 2009 Jun 16:72(24):2083-9. doi: 10.1212/WNL.0b013e3181aa5364. Epub [PubMed PMID: 19528514]
Tiryaki E, Horak HA. ALS and other motor neuron diseases. Continuum (Minneapolis, Minn.). 2014 Oct:20(5 Peripheral Nervous System Disorders):1185-207. doi: 10.1212/01.CON.0000455886.14298.a4. Epub [PubMed PMID: 25299277]
Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet (London, England). 2011 Mar 12:377(9769):942-55. doi: 10.1016/S0140-6736(10)61156-7. Epub 2011 Feb 4 [PubMed PMID: 21296405]
Limousin N, Blasco H, Corcia P, Gordon PH, De Toffol B, Andres C, Praline J. Malnutrition at the time of diagnosis is associated with a shorter disease duration in ALS. Journal of the neurological sciences. 2010 Oct 15:297(1-2):36-9. doi: 10.1016/j.jns.2010.06.028. Epub 2010 Jul 31 [PubMed PMID: 20673675]
Level 2 (mid-level) evidenceWesteneng HJ, Debray TPA, Visser AE, van Eijk RPA, Rooney JPK, Calvo A, Martin S, McDermott CJ, Thompson AG, Pinto S, Kobeleva X, Rosenbohm A, Stubendorff B, Sommer H, Middelkoop BM, Dekker AM, van Vugt JJFA, van Rheenen W, Vajda A, Heverin M, Kazoka M, Hollinger H, Gromicho M, Körner S, Ringer TM, Rödiger A, Gunkel A, Shaw CE, Bredenoord AL, van Es MA, Corcia P, Couratier P, Weber M, Grosskreutz J, Ludolph AC, Petri S, de Carvalho M, Van Damme P, Talbot K, Turner MR, Shaw PJ, Al-Chalabi A, Chiò A, Hardiman O, Moons KGM, Veldink JH, van den Berg LH. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. The Lancet. Neurology. 2018 May:17(5):423-433. doi: 10.1016/S1474-4422(18)30089-9. Epub 2018 Mar 26 [PubMed PMID: 29598923]
Level 1 (high-level) evidenceMajmudar S, Wu J, Paganoni S. Rehabilitation in amyotrophic lateral sclerosis: why it matters. Muscle & nerve. 2014 Jul:50(1):4-13. doi: 10.1002/mus.24202. Epub 2014 May 17 [PubMed PMID: 24510737]
Level 3 (low-level) evidencePaganoni S, Karam C, Joyce N, Bedlack R, Carter GT. Comprehensive rehabilitative care across the spectrum of amyotrophic lateral sclerosis. NeuroRehabilitation. 2015:37(1):53-68. doi: 10.3233/NRE-151240. Epub [PubMed PMID: 26409693]
Chiò A, Bottacchi E, Buffa C, Mutani R, Mora G, PARALS. Positive effects of tertiary centres for amyotrophic lateral sclerosis on outcome and use of hospital facilities. Journal of neurology, neurosurgery, and psychiatry. 2006 Aug:77(8):948-50 [PubMed PMID: 16614011]
Level 2 (mid-level) evidenceTraynor BJ, Alexander M, Corr B, Frost E, Hardiman O. Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996-2000. Journal of neurology, neurosurgery, and psychiatry. 2003 Sep:74(9):1258-61 [PubMed PMID: 12933930]
Fidelix EC, Santana GC, Barros DMDS, Dourado Junior MET. Telehealth for amyotrophic lateral sclerosis in a multidisciplinary service in a Brazilian reference center. Arquivos de neuro-psiquiatria. 2023 May:81(5):469-474. doi: 10.1055/s-0043-1768161. Epub 2023 May 31 [PubMed PMID: 37257467]