
Introduction
Achondroplasia is a rare genetic disorder recognized as the most common primary skeletal dysplasia in humans. This form of dysplasia accounts for greater than 90% of cases of disproportionate short stature, also known as dwarfism.[1] The term “achondroplasia” was first used in 1878 to distinguish it from rickets, one of many other abnormal conditions of bone growth.
Achondroplasia means “without cartilage formation,” and it is categorized as a physeal (growth plate) dysplasia.[2] The condition’s causative mutation of the transmembrane portion of fibroblast growth factor receptor 3 (FGFR3) was discovered in 1995.[3] It follows an autosomal dominant pattern of inheritance with 100% penetrance. Over 80% of cases arise from a spontaneous mutation, and advanced paternal age is a known risk factor. The phenotype is distinct from other skeletal dysplasias and can be identified on prenatal ultrasonography and by newborn examination.
Affected individuals usually present average intelligence and have an estimated mean lifespan of 61 years, about ten years less than the general population.[4] Physical phenotypic features include large head size (macrocephaly) with frontal bossing, midface hypoplasia, rhizomelic shortening of the extremities, short fingers (brachydactyly) with trident configuration of the hand, and bowed legs (genu varum).
Achondroplasia is associated with increased mortality in early childhood, otolaryngology problems later in childhood, and increased risk of obesity into adulthood.[4] Affected individuals can also develop joint laxity, thoracolumbar kyphosis (TLK), and spinal stenosis that may progress and contribute to morbidity as an adult.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Achondroplasia results from a point mutation in the gene coding for the transmembrane portion of fibroblast growth factor receptor 3 (FGFR3), which resides on the short arm of chromosome 4.[5] The resultant abnormal chondroid production affects endochondral ossification, resulting in decreased linear bone growth, among other functions. This pathologic process generally spares intramembranous ossification, which takes place in flat bones such as those in the skull (with the exception of the base of the skull), face, and clavicles.
- In over 80% of cases, the condition occurs due to sporadic, or de novo, mutation. Thus, a child with achondroplasia can be born to healthy parents with no family history of the disorder.
- The remaining 20% of achondroplastic individuals have at least one affected parent.
- Achondroplasia is inherited in an autosomal dominant manner. It is fully penetrant, meaning all individuals who have the FGFR3 heterozygous pathogenic variant show the clinical manifestations of the disorder.[2]
- The “unprecedented” homogeneity of mutations of this autosomal dominant disorder leads to the relative lack of heterogeneity in the achondroplasia phenotype.[3]
- The risk to offspring of an achondroplastic individual of inheriting a mutated copy of the FGFR3 gene is 50%. When both parents are affected, their offspring have a 1 in 4 (25%) chance of having normal stature, a 1 in 2 (50%) chance at having achondroplasia (heterozygous), and a 1 in 4 (25%) chance of homozygous achondroplasia.[2]
- The homozygous form is usually incompatible with life, usually resulting in early neonatal death from respiratory insufficiency due to a small thoracic cage and neurologic deficits from cervicomedullary stenosis.[6]
One well-known risk factor for producing offspring with the de novo mutation is advanced paternal age, with the mutation thought to occur during spermatogenesis. An increased risk of achondroplasia is not associated with older maternal age, independently of the older paternal ages.[7]
Epidemiology
Achondroplasia occurs in approximately 1:20,000 to 1:30,000 live births per year.[7][8][9] The global prevalence is difficult to ascertain; however, it is estimated to affect approximately 1 to 9 individuals per 100,000 of the general population. The most extensive European population-based epidemiological study known to date calculated the prevalence to be 3.72 per 100,000 births.[10] The study demonstrated prevalence was stable over time, yet regional differences were appreciated. As achondroplasia follows an autosomal dominant inheritance pattern, both males and females are equally affected.
The estimated occurrence rate for this form of skeletal dysplasia in children of fathers over 50 years of age is 1 in 1,875, much higher when compared to the frequency of approximately 1 in 15,000 in the general population.[9][10][11][12] Fathers with ages 35 or older had significantly increased rates of affected offspring.
Pathophysiology
A point mutation in the gene coding for the transmembrane portion of FGFR3, which resides on the short arm of chromosome 4, results in achondroplasia.
The point mutation arises from two possible base substitutions: a transition of c.1138G>A (guanine to adenine substitution is identified in approximately 98% of affected individuals) and a transversion of c.1138G>C (guanine to cytosine, seen in about 1% of affected individuals).[8] These base substitutions cause the normal GGG codon to change to AGG or CGG, causing glycine to be replaced with arginine (p.Gly380Arg) in both situations, and subsequently affecting the transmembrane domain of FGFR3. Both substitutions confer a pathogenic variant of FGFR3, which leads to a gain-of-function mechanism of FGFR3 and subsequent quantitative growth plate and cartilage defects seen in achondroplasia.
GFR3 normal function is to slow the formation of bone by inhibiting the proliferation of chondrocytes in the proliferative zone of the physis of long bones.[13]
The genetic mutation of FGFR3 (p.Gly380Arg) results in a gain-of-function, constitutive activation of the receptor protein, and a significant decrease in endochondral bone formation via increased inhibition of chondrocyte proliferation and differentiation.[14] This process results in the clinical phenotypic features.
There is associated increased mortality in childhood, likely due to FMS. The stenosis at the base of the skull can cause cervicomedullary myelopathy (compression of a portion of the brainstem and spinal cord that may cause central sleep apnea, difficulty walking, difficulty swallowing, weakness, numbness, and loss of bowel or bladder control). The average adult height in achondroplasia is approximately 4 feet for both sexes.[15]
History and Physical
Achondroplasia is characterized by short stature, macrocephaly with frontal bossing (broad forehead), midface hypoplasia (small nasal bridge), FMS (the base of the skull is endochondral in origin), rhizomelia (the proximal portion of the limb is shorter than distal portion), brachydactyly (short digits) with a prominent gap between the ring and middle fingers (known as a "trident hand"), radial head subluxation, posterior bowing of the humerus, thoracolumbar kyphosis (TLK), lumbar hyperlordosis, and genu varum (bowed legs).[15][16]
Those affected have an average torso size, yet short limbs (sitting height may be within normal limits), but their standing height will often be below the 5th percentile. The upper extremities may not be able to extend at the elbows fully. An achondroplastic cohort's upper extremity range of motion was investigated and revealed a mean loss of elbow extension of approximately 13.1 degrees, primarily due to posterior bowing of the distal humerus and less so due to posterior radial head subluxations.[17]
Bowed legs are very common in those with achondroplasia. The technical term for this bowing is genu varum, and more than 90% of untreated adults with achondroplasia have some degree of bowing.[18] This deformity is structurally complex, originating from a combination of internal tibial torsion, lateral bowing, and some dynamic instability and laxity of the knee.[19]
Thoracolumbar kyphosis (TLK) is often appreciated in the newborn and can be accentuated due to massive head size, trunk hypotonia (thought to be the etiology of this feature), flat chest, and protuberant abdomen, especially when the infant begins to sit upright. The visible deformity may correct when the child lays prone. T12 and L1 apical wedging are frequently seen on x-rays, and the deformity usually involves vertebrae of T10 to L4. The frequency of TLK deformity was found to be as high as 87% at 1 to 2 years old and decreased to 11% from 5 to 10 years old.[20] Radiographic predictors of persistent TLK include apical vertebral translation (seen in 5% of those affected) and apical vertebral wedging for vertebral height (seen in 6% of those affected).[21] Developmental motor delay is also associated with unresolved TLK. It is recommended that patients with kyphotic curves between 20 and 40 degrees be monitored for progressive deformity and symptoms of spinal cord compression. As the child begins to stand and walk, lumbosacral hyperlordosis may become apparent. This additional deformity is likely due to excessive anterior pelvic tilt in these children while standing.
Foramen magnum stenosis (FMS) is usually the first spinal manifestation observed in infants with achondroplasia. Increased vigilance is required in both the evaluation and surveillance of this condition. It is associated with a high mortality rate, ranging from 2 to 5%. Common signs and symptoms include periods of apnea while sleeping and excessive snoring. Other subjective or objective findings include difficulty swallowing, lower cranial nerve palsies, hyperreflexia, generalized hypotonia, weakness, and clonus. Intelligence is usually average unless the child develops hydrocephalus or has other central nervous system complications.[16] Some infants will also develop hydrocephalus, which is thought to be due to increased intracranial venous pressure secondary to stenosis of the jugular foramina. The frequency of hydrocephalus requiring treatment is estimated to be 5% or less of those affected.[22]
Hearing impairment can be observed in children with achondroplasia as a result of middle ear dysfunction. If inadequately treated, these problems can result in conductive hearing loss, which may be severe enough to interfere with language development. Approximately 50% of children will require a pressure-equalizing tympanostomy and ear tube placement.[23] About 40% of achondroplastic patients have functionally relevant hearing loss.[24]
Pseudoclaudication, discomfort while standing, and difficulty walking for prolonged periods, leg paresthesias, and subjective weakness may be observed later in development. These are specific signs of spinal stenosis, and patients with achondroplasia are prone to lumbar stenosis due to the development of short pedicles, thickened facets, and thickened ligamentum flavum. Lumbar spinal stenosis occurs in approximately 25% of achondroplastic patients. Neurologic symptoms such as lower extremity numbness are usually related to nerve root or spinal cord compression.
Evaluation
There are no diagnostic algorithms for achondroplasia; the diagnosis can usually be made with certain characteristics and specific radiologic features.
- Diagnosis can be confirmed by molecular genetic testing for the FGFR3 mutation. FGFR3 testing is indicated in children with an atypical presentation or to differentiate from similar disorders. A multigene panel, including FGFR3 and other genes of interest, can be performed to help ascertain the specific form of dysplasia.
- Prenatal diagnosis can occur incidentally during a routine second or third-trimester ultrasound if shortened long bones are appreciated in the developing fetus. Noninvasive prenatal diagnosis using cell-free fetal DNA found in the mother’s serum is now available, with reported high sensitivity and specificity.[1][8]
- Preimplantation genetic diagnosis is also now possible for parents pursuing in-vitro fertilization and embryo implantation procedures. The prenatal detection rate is significantly higher in recent years; 71% during 2011-2015 compared to 36% during 1991-1995.[10]
- Skeletal surveys of achondroplastic children may demonstrate a contracted skull base, rhizomelic shortening of long bones, proximal femoral radiolucency, generalized metaphyseal “flaring” irregularities, inverted “V-shaped or chevron-shaped” distal femoral epiphyses, a “champagne-glass” shaped pelvis (wider than deep pelvic outlet with small sacrosciatic notch). The radiographic appearance of the spine also differs in those with achondroplasia, including narrowed interpedicular distances (short pedicles usually found from L1-S1), vertebral body wedging (usually found at T12 or L1) and generalized posterior vertebral scalloping.
- If there is evidence of sleep apnea in the affected individual, or development of apparent signs and symptoms of cervicomedullary myelopathy, a sleep study and advanced imaging of the cervicomedullary junction may be indicated such as a computed tomographic (CT) scan or magnetic resonance imaging (MRI).
Treatment / Management
Management of achondroplasia involves an interprofessional team approach, and anticipatory care is essential. Multiple versions of health supervision and treatment guidelines exist for those with achondroplasia, with most including detailed examinations and specific anticipatory guidance for each separate age group.[25] These guidelines should be followed closely by the pediatrician and/or other health practitioners. Close attention should be paid to the development of obesity in these children and monitored to avoid complications later in life. Treatment is directed against the specific issues encountered in achondroplasia.(B2)
Macrocephaly
- It is crucial to distinguish between a “normal” amount of macrocephaly (large ventricles under normal pressure) and clinically significant hydrocephalus. Therefore, head circumference measurements should be performed at every health care contact and plotted on achondroplasia-specific standards for surveillance.[15]
- Due to the delay in skull sutural maturation in achondroplasia, measurements of cranial circumference should continue until six years of age. Hydrocephalus requiring treatment occurs in approximately 5%.[22]
- The foramen magnum stenosis may contribute to significant hydrocephalus in these children. This can be surgically corrected by endoscopic third ventriculostomy, ventricular shunt, or by posterior fossa decompression.[26] (B3)
Foramen Magnum Stenosis
- For those patients with FMS, the American Academy of Pediatrics recommends obtaining a sleep study (polysomnography) first, and then if positive, obtaining a CT scan or MRI to evaluate the cervicomedullary area for compressive myelopathy. If either study returns abnormal, prompt evaluation by neurosurgery should be arranged for possible surgical treatment.
- MRIs in those with FMS will likely show a relatively large cranial vault with a small, contracted appearing skull base (in addition to the narrowed foramen magnum), with a prominent forehead and depressed nasal bridge. There may be visible “cervicomedullary kink” in those with severe FMS.
- The condition is usually treated in the first two years of life. However, affected patients may become symptomatic later in life.
- Treatment is surgical with foramen magnum decompression and upper cervical laminectomy with or without duraplasty.[27]
- Complete resolution or partial improvement in the preoperative symptoms is usually obtained. Children with FMS identified on imaging studies should avoid activities that place them at risk of head or neck injury. (B2)
Hearing impairment
- Referral to a pediatric otolaryngologist should be obtained for those who develop frequent otitis media and/or hearing problems. Long-lasting tympanostomy tubes may be necessary and are frequently required until the age of 8 years.
- In addition to newborn screening, infants should have an audiometric evaluation by one year of age. Audiology evaluations should be performed each year until school age and should continue on an annual basis if tympanic tubes were placed.
- Speech therapy should also be offered if there are concerns about hearing loss.
- Obstructive sleep apnea (OSA) from a non-central etiology needs to be ruled out and treated by a pediatric otolaryngologist specialist. Children with severe OSA may require weight reduction, adenotonsillectomy, positive airway pressure at night, and/or tracheostomy for very severe cases.
Thoracolumbar Kyphosis (TLK)
- TLK often presents in the newborn and usually resolves spontaneously by 18 months of age in approximately 90% of the affected children, paralleling the child’s ability to ambulate as the trunk muscles strengthen.[28]
- It is recommended that unsupported sitting should be limited in achondroplastic children less than one year of age to help prevent the progression of kyphosis.
- Bracing may be indicated if TLK progresses to a fixed deformity > 30 degrees (measured when prone) and when there is anterior vertebral wedging or posterior displacement of vertebrae at the apex of deformity.
- Continuation of bracing is recommended until the child is walking independently, the anterior corners of the vertebrae reconstitute, and the fixed component of the curve shows no further improvement. As bracing in these children may be poorly tolerated, an anterior and posterior fusion can be performed for a residual sagittal deformity greater than 60 degrees by age 5.
- Surgery should be reserved for those with neurologic deficits or fixed kyphosis measuring > 50 degrees.[13] TLK can improve alone before the third year.[29] A compensatory mechanism of hyperlordosis at the lumbosacral level after age 3 plays action as TLK is shown to decrease gradually up to the age of 10.
- Treatment indications of the hyperlordosis are poorly defined, usually consisting of nonoperative modalities and observation throughout childhood. (A1)
Lumbar Spinal Stenosis
- Lumbar spinal stenosis is the most likely cause of disability later in life. Evaluation for stenosis with x-rays should be considered in all patients wishing to play contact sports or participate in high-risk activities.
- Lumbar stenosis may first be treated nonoperatively with modalities such as weight loss, physical therapy, and corticosteroid injections.
- Operative intervention is indicated when there are progressive symptoms such as severe claudication, urinary retention, and/or neurologic symptoms while at rest.
- Surgical treatment includes multi-level laminectomy, including lateral recess decompression with instrumented posterior spinal fusion to avoid post-laminectomy kyphotic deformity.[30] (B2)
Bowed Legs
- Examination for bowing and/or internal tibial torsion must be part of each physical assessment of these children, with an immediate orthopedic referral for any concerns.
- It is often difficult to define indications for surgery in these children, but they may include the onset of knee or leg pain, changes in ambulation (lateral thrust), and/or severe alignment.
- Those with progressive and symptomatic leg bowing can benefit from either hemiepiphysiodesis or tibial/fibular osteotomies with or without femoral osteotomies.
- Options for limb lengthening include callodiastasis (lengthening through a metaphyseal corticotomy). However, these procedures are controversial due to high rates of complications.[31]
Pharmacological Therapy: There are a few ongoing clinical trials. One therapeutic drug called vosoritide, a biologic analog of a potent stimulator of endochondral ossification, recently cleared Phase III of its clinical trial with promising results.[32][33][34] The drug demonstrated a placebo-adjusted increase in growth velocity of 1.6cm/yr (p<0.0001) in achondroplastic children. Treatment with once-daily subcutaneous administration of vosoritide resulted in a sustained increase in the annualized growth velocity for up to 42 months measured, with a generally mild side-effect profile.
Differential Diagnosis
There are over 350 skeletal dysplasias known to cause short stature, most of which are very rare.[35] However, only a handful of conditions exist that may be confused with achondroplasia. Hypochondroplasia and thanatophoric dysplasia are two other disorders with features of rhizomelic dwarfism. Both arise from a similar genetic defect as achondroplasia, with different pathogenic variants in FGFR3 that result in differing levels of FGFR3 activation, and both should be included in the differential diagnosis.[1] FGFR3 molecular genetic testing should always be performed in children with an atypical presentation or in circumstances in which differentiation is required from similar disorders.
The distinction between achondroplasia and hypochondroplasia is usually very difficult to make as there is overlap between most of the radiologic and clinical features of the two conditions.[36] Patients affected by this disorder appear normal at birth. However, their arms and legs do not develop properly, and their body becomes thicker and shorter than normal. It is caused by a defect in the FGFR3 at chromosome 4 at the 4p16.3 area. There is less height difference than in achondroplasia. Approximately 10% of cases have mild mental retardation.
Thanatophoric dysplasia (TD) causes a form of short-limb dwarfism and is typically lethal in the perinatal period. TD has two clinically distinct forms, type I and type II. Type I is characterized by micromelia with bowed femurs. Type II is characterized by micromelia with straight femurs and uniform presence of moderate-to-severe cloverleaf skull deformity (Kleeblattschädel). Varying severity of the cloverleaf skull can also be present in type I. Both types have features in common, including infantile hypotonia, macrocephaly, frontal bossing, flat facies with ocular proptosis, brachydactyly, and micromelia. Most infants with this disorder die due to respiratory insufficiency shortly following birth.[37] The condition is usually incidentally discovered and suspected during a routine prenatal ultrasound, with characteristic findings of shortened long bones visible as early as 14 weeks gestation. Specific findings during second and third-trimester ultrasounds include a cloverleaf skull, macrocephaly, ventriculomegaly, increased nuchal translucency, a narrow chest cavity with short ribs, and bowed femurs (TD type I). TD is also only caused by mutations of the FGFR3 gene and the pathogenic variant p.Lys650Glu has been identified in all diagnosed individuals with TD type II.[38]
Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) is another dysplastic condition that arises from a different heterozygous mutation in the gene encoding FGFR3 on chromosome 4p16. These individuals can develop extensive areas of acanthosis nigricans starting in early childhood and suffer from severe neurological impairments.[39] Investigations of SADDAN revealed that the mutation resulted in severe disturbances in endochondral bone growth, which approached the severity seen in thanatophoric dysplasia type I (which is generally not compatible with survival into adulthood). Osseous deformities seen included femoral bowing, apex posterior tibial, and fibular bowing and curved “ram’s horn” deformities of the clavicles.[40] Acanthosis nigricans can also be seen in those with other FGFR3 genetic disorders. Therefore the progressive skin changes should be thought of as a long-term complication rather than a specific feature of SADDAN.[41]
Pseudoachondroplasia, a clinically and genetically distinct skeletal dysplasia, may also cause confusion. This condition also follows an autosomal dominant pattern of inheritance yet is due to a defect of cartilage oligomeric matrix protein, found on chromosome 19. Signs and symptoms of this disorder include cervical instability and scoliosis with increased lumbar lordosis and significant lower extremity bowing. Individuals with this condition can acquire hip, knee, and elbow flexion contractures with precocious osteoarthritis. Radiographic findings include metaphyseal flaring and delayed epiphyseal ossification.[42]
Prognosis
Those diagnosed with this disorder have increased mortality rates in childhood. Multiple studies have demonstrated that the osseous abnormalities were seen in achondroplastic patients, such as FMS and spinal stenosis, contribute to the significant effect on morbidity and mortality seen in all age groups, but mainly affect the pediatric group who present with FMS. The mean lifespan of patients with achondroplasia has been estimated to be 61 years, approximately ten years shorter when compared to the general population.[4]
Complications
There are many medical complications associated with a diagnosis of achondroplasia. These include but are not limited to hydrocephalus, otolaryngology problems (recurrent otitis media, hearing loss, obstructive sleep apnea), rapid infantile weight velocity, and obesity during childhood. Surgical complications can also lead to morbidity.
Consultations
As a result of its wide spectrum of manifestations, it is vital to include an interprofessional team, including a pediatrician, orthopedic surgeon, neurosurgeon, otolaryngologist, pneumologist, sleep/apnea specialist, neurologist, physiatrist, and a geneticist for the management of the disease.
Deterrence and Patient Education
Achondroplasia accounts for greater than 90% of cases of dwarfism. Although it follows an autosomal dominant pattern of inheritance with a 100% penetrant, over 80% of cases arise from a spontaneous mutation. Genetic counseling is recommended for patients. It is associated with increased mortality in early childhood, otolaryngology problems later in childhood, and increased risk of obesity into adulthood.
There are many medical complications associated with a diagnosis of achondroplasia. Increased vigilance is required in both the evaluation and surveillance of this condition. Affected individuals can also develop joint laxity, thoracolumbar kyphosis, and spinal stenosis that may progress and contribute to morbidity as an adult. FMS is usually the first spinal manifestation observed in infants with achondroplasia. Lumbar spinal stenosis occurs in approximately 25% of achondroplastic patients. Management of achondroplasia involves a multidisciplinary team approach and anticipatory care is essential. The mean lifespan of patients with achondroplasia is slightly shorter than the average population.
Enhancing Healthcare Team Outcomes
The management of a child with achondroplasia can be enhanced by utilizing a multidisciplinary team approach. It should involve pediatricians, geneticists, pediatric neurologists, pediatric pulmonologists, pediatric neurosurgeons, and pediatric orthopedic surgeons[43] Anticipatory care is essential, and efforts should be directed at identifying affected children who are at high risk and appropriately intervening to prevent permanent sequelae. Due to the extensive list of possible comorbidities that these individuals may face in their lifetime, such as obesity and lumbar spinal stenosis, evaluation and treatment by a physical therapist may be beneficial with an emphasis on preventative therapies and strict adherence to home exercises. Because of the apparent nature of the short stature, those affected and their families may encounter difficulties in school adjustment and socialization. Formal support groups can assist families with these concerns through peer support and social awareness programs.[8] Many of these organizations have national conferences each year. There are also many resources available for information on disability rights, employment, medical issues, adaptive devices, etc. through seminars, workshops, and national newsletters.
Media
(Click Image to Enlarge)
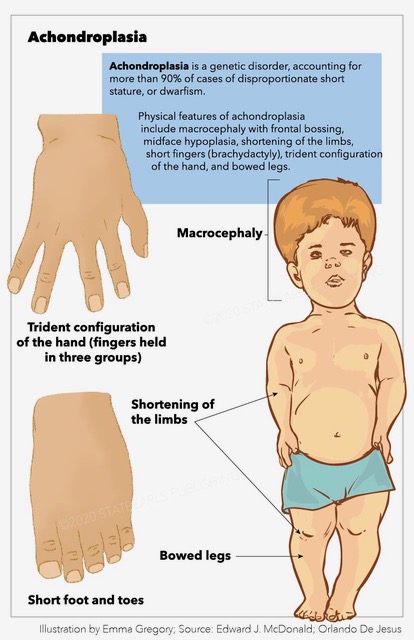
Achondroplasia, trident configuration, macrocephaly, bowed legs, dwarfism, frontal bossing
Contributed by StatPearls Publishing Illustration
References
Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocrine reviews. 2000 Feb:21(1):23-39 [PubMed PMID: 10696568]
Wang X, Ramström O, Yan M. Glyconanomaterials: synthesis, characterization, and ligand presentation. Advanced materials (Deerfield Beach, Fla.). 2010 May 4:22(17):1946-53. doi: 10.1002/adma.200903908. Epub [PubMed PMID: 20301131]
Bellus GA, Hefferon TW, Ortiz de Luna RI, Hecht JT, Horton WA, Machado M, Kaitila I, McIntosh I, Francomano CA. Achondroplasia is defined by recurrent G380R mutations of FGFR3. American journal of human genetics. 1995 Feb:56(2):368-73 [PubMed PMID: 7847369]
Hecht JT, Francomano CA, Horton WA, Annegers JF. Mortality in achondroplasia. American journal of human genetics. 1987 Sep:41(3):454-64 [PubMed PMID: 3631079]
Horton WA. Recent milestones in achondroplasia research. American journal of medical genetics. Part A. 2006 Jan 15:140(2):166-9 [PubMed PMID: 16353253]
Level 3 (low-level) evidenceHall JG. The natural history of achondroplasia. Basic life sciences. 1988:48():3-9 [PubMed PMID: 3071358]
Waller DK, Correa A, Vo TM, Wang Y, Hobbs C, Langlois PH, Pearson K, Romitti PA, Shaw GM, Hecht JT. The population-based prevalence of achondroplasia and thanatophoric dysplasia in selected regions of the US. American journal of medical genetics. Part A. 2008 Sep 15:146A(18):2385-9. doi: 10.1002/ajmg.a.32485. Epub [PubMed PMID: 18698630]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Legare JM. Achondroplasia. GeneReviews(®). 1993:(): [PubMed PMID: 20301331]
Oberklaid F, Danks DM, Jensen F, Stace L, Rosshandler S. Achondroplasia and hypochondroplasia. Comments on frequency, mutation rate, and radiological features in skull and spine. Journal of medical genetics. 1979 Apr:16(2):140-6 [PubMed PMID: 458831]
Level 3 (low-level) evidenceCoi A, Santoro M, Garne E, Pierini A, Addor MC, Alessandri JL, Bergman JEH, Bianchi F, Boban L, Braz P, Cavero-Carbonell C, Gatt M, Haeusler M, Klungsøyr K, Kurinczuk JJ, Lanzoni M, Lelong N, Luyt K, Mokoroa O, Mullaney C, Nelen V, Neville AJ, O'Mahony MT, Perthus I, Rankin J, Rissmann A, Rouget F, Schaub B, Tucker D, Wellesley D, Wisniewska K, Zymak-Zakutnia N, Barišić I. Epidemiology of achondroplasia: A population-based study in Europe. American journal of medical genetics. Part A. 2019 Sep:179(9):1791-1798. doi: 10.1002/ajmg.a.61289. Epub 2019 Jul 11 [PubMed PMID: 31294928]
Kovac JR, Addai J, Smith RP, Coward RM, Lamb DJ, Lipshultz LI. The effects of advanced paternal age on fertility. Asian journal of andrology. 2013 Nov:15(6):723-8. doi: 10.1038/aja.2013.92. Epub 2013 Aug 5 [PubMed PMID: 23912310]
Wyrobek AJ, Eskenazi B, Young S, Arnheim N, Tiemann-Boege I, Jabs EW, Glaser RL, Pearson FS, Evenson D. Advancing age has differential effects on DNA damage, chromatin integrity, gene mutations, and aneuploidies in sperm. Proceedings of the National Academy of Sciences of the United States of America. 2006 Jun 20:103(25):9601-6 [PubMed PMID: 16766665]
Shirley ED, Ain MC. Achondroplasia: manifestations and treatment. The Journal of the American Academy of Orthopaedic Surgeons. 2009 Apr:17(4):231-41 [PubMed PMID: 19307672]
Laederich MB, Horton WA. Achondroplasia: pathogenesis and implications for future treatment. Current opinion in pediatrics. 2010 Aug:22(4):516-23. doi: 10.1097/MOP.0b013e32833b7a69. Epub [PubMed PMID: 20601886]
Level 3 (low-level) evidenceHorton WA, Rotter JI, Rimoin DL, Scott CI, Hall JG. Standard growth curves for achondroplasia. The Journal of pediatrics. 1978 Sep:93(3):435-8 [PubMed PMID: 690757]
Wigg K, Tofts L, Benson S, Porter M. The neuropsychological function of children with achondroplasia. American journal of medical genetics. Part A. 2016 Nov:170(11):2882-2888. doi: 10.1002/ajmg.a.37779. Epub 2016 Sep 8 [PubMed PMID: 27605460]
Kitoh H, Kitakoji T, Kurita K, Katoh M, Takamine Y. Deformities of the elbow in achondroplasia. The Journal of bone and joint surgery. British volume. 2002 Jul:84(5):680-3 [PubMed PMID: 12188484]
Kopits SE. Genetics clinics of The Johns Hopkins Hospital. Surgical intervention in achondroplasia. Correction of bowleg deformity in achondroplasia. The Johns Hopkins medical journal. 1980 May:146(5):206-9 [PubMed PMID: 7382244]
Level 3 (low-level) evidenceInan M, Thacker M, Church C, Miller F, Mackenzie WG, Conklin D. Dynamic lower extremity alignment in children with achondroplasia. Journal of pediatric orthopedics. 2006 Jul-Aug:26(4):526-9 [PubMed PMID: 16791073]
Level 2 (mid-level) evidenceKopits SE. Thoracolumbar kyphosis and lumbosacral hyperlordosis in achondroplastic children. Basic life sciences. 1988:48():241-55 [PubMed PMID: 3240259]
Margalit A, McKean G, Lawing C, Galey S, Ain MC. Walking Out of the Curve: Thoracolumbar Kyphosis in Achondroplasia. Journal of pediatric orthopedics. 2018 Nov/Dec:38(10):491-497. doi: 10.1097/BPO.0000000000000862. Epub [PubMed PMID: 27636912]
Steinbok P, Hall J, Flodmark O. Hydrocephalus in achondroplasia: the possible role of intracranial venous hypertension. Journal of neurosurgery. 1989 Jul:71(1):42-8 [PubMed PMID: 2786928]
Level 3 (low-level) evidenceBerkowitz RG, Grundfast KM, Scott C, Saal H, Stern H, Rosenbaum K. Middle ear disease in childhood achondroplasia. Ear, nose, & throat journal. 1991 May:70(5):305-8 [PubMed PMID: 1914954]
Level 2 (mid-level) evidenceIreland PJ, Donaghey S, McGill J, Zankl A, Ware RS, Pacey V, Ault J, Savarirayan R, Sillence D, Thompson E, Townshend S, Johnston LM. Development in children with achondroplasia: a prospective clinical cohort study. Developmental medicine and child neurology. 2012 Jun:54(6):532-7. doi: 10.1111/j.1469-8749.2012.04234.x. Epub 2012 Mar 12 [PubMed PMID: 22409389]
Level 2 (mid-level) evidenceHoover-Fong JE, McGready J, Schulze KJ, Barnes H, Scott CI. Weight for age charts for children with achondroplasia. American journal of medical genetics. Part A. 2007 Oct 1:143A(19):2227-35 [PubMed PMID: 17764078]
Level 2 (mid-level) evidenceJiang ZH. [Conray ventriculography in the diagnosis of tumors in the posterior cranial fossa (author's transl)]. Zhonghua fang she xue za zhi Chinese journal of radiology. 1979:13(2):84-7 [PubMed PMID: 162380]
Bagley CA, Pindrik JA, Bookland MJ, Camara-Quintana JQ, Carson BS. Cervicomedullary decompression for foramen magnum stenosis in achondroplasia. Journal of neurosurgery. 2006 Mar:104(3 Suppl):166-72 [PubMed PMID: 16572633]
Level 2 (mid-level) evidenceEngberts AC, Jacobs WC, Castelijns SJ, Castelein RM, Vleggeert-Lankamp CL. The prevalence of thoracolumbar kyphosis in achondroplasia: a systematic review. Journal of children's orthopaedics. 2012 Mar:6(1):69-73. doi: 10.1007/s11832-011-0378-7. Epub 2011 Dec 3 [PubMed PMID: 22442656]
Level 1 (high-level) evidenceAbousamra O, Shah SA, Heydemann JA, Kreitz TM, Rogers KJ, Ditro C, Mackenzie WG. Sagittal Spinopelvic Parameters in Children With Achondroplasia. Spine deformity. 2019 Jan:7(1):163-170. doi: 10.1016/j.jspd.2018.06.001. Epub [PubMed PMID: 30587311]
Ain MC, Shirley ED, Pirouzmanesh A, Hariri A, Carson BS. Postlaminectomy kyphosis in the skeletally immature achondroplast. Spine. 2006 Jan 15:31(2):197-201 [PubMed PMID: 16418640]
Level 2 (mid-level) evidenceMonticelli G, Spinelli R. Leg lengthening by closed metaphyseal corticotomy. Italian journal of orthopaedics and traumatology. 1983 Jun:9(2):139-50 [PubMed PMID: 6654651]
Semler O, Rehberg M, Mehdiani N, Jackels M, Hoyer-Kuhn H. Current and Emerging Therapeutic Options for the Management of Rare Skeletal Diseases. Paediatric drugs. 2019 Apr:21(2):95-106. doi: 10.1007/s40272-019-00330-0. Epub [PubMed PMID: 30941653]
Breinholt VM, Rasmussen CE, Mygind PH, Kjelgaard-Hansen M, Faltinger F, Bernhard A, Zettler J, Hersel U. TransCon CNP, a Sustained-Release C-Type Natriuretic Peptide Prodrug, a Potentially Safe and Efficacious New Therapeutic Modality for the Treatment of Comorbidities Associated with Fibroblast Growth Factor Receptor 3-Related Skeletal Dysplasias. The Journal of pharmacology and experimental therapeutics. 2019 Sep:370(3):459-471. doi: 10.1124/jpet.119.258251. Epub 2019 Jun 24 [PubMed PMID: 31235532]
Savarirayan R, Irving M, Bacino CA, Bostwick B, Charrow J, Cormier-Daire V, Le Quan Sang KH, Dickson P, Harmatz P, Phillips J, Owen N, Cherukuri A, Jayaram K, Jeha GS, Larimore K, Chan ML, Huntsman Labed A, Day J, Hoover-Fong J. C-Type Natriuretic Peptide Analogue Therapy in Children with Achondroplasia. The New England journal of medicine. 2019 Jul 4:381(1):25-35. doi: 10.1056/NEJMoa1813446. Epub 2019 Jun 18 [PubMed PMID: 31269546]
Krakow D, Rimoin DL. The skeletal dysplasias. Genetics in medicine : official journal of the American College of Medical Genetics. 2010 Jun:12(6):327-41. doi: 10.1097/GIM.0b013e3181daae9b. Epub [PubMed PMID: 20556869]
Almeida MR, Campos-Xavier AB, Medeira A, Cordeiro I, Sousa AB, Lima M, Soares G, Rocha M, Saraiva J, Ramos L, Sousa S, Marcelino JP, Correia A, Santos HG. Clinical and molecular diagnosis of the skeletal dysplasias associated with mutations in the gene encoding Fibroblast Growth Factor Receptor 3 (FGFR3) in Portugal. Clinical genetics. 2009 Feb:75(2):150-6. doi: 10.1111/j.1399-0004.2008.01123.x. Epub [PubMed PMID: 19215249]
Level 3 (low-level) evidenceAdam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, French T, Savarirayan R. Thanatophoric Dysplasia. GeneReviews(®). 1993:(): [PubMed PMID: 20301540]
Bellus GA, Spector EB, Speiser PW, Weaver CA, Garber AT, Bryke CR, Israel J, Rosengren SS, Webster MK, Donoghue DJ, Francomano CA. Distinct missense mutations of the FGFR3 lys650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia phenotype. American journal of human genetics. 2000 Dec:67(6):1411-21 [PubMed PMID: 11055896]
Level 3 (low-level) evidenceTavormina PL, Bellus GA, Webster MK, Bamshad MJ, Fraley AE, McIntosh I, Szabo J, Jiang W, Jabs EW, Wilcox WR, Wasmuth JJ, Donoghue DJ, Thompson LM, Francomano CA. A novel skeletal dysplasia with developmental delay and acanthosis nigricans is caused by a Lys650Met mutation in the fibroblast growth factor receptor 3 gene. American journal of human genetics. 1999 Mar:64(3):722-31 [PubMed PMID: 10053006]
Bellus GA, Bamshad MJ, Przylepa KA, Dorst J, Lee RR, Hurko O, Jabs EW, Curry CJ, Wilcox WR, Lachman RS, Rimoin DL, Francomano CA. Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN): phenotypic analysis of a new skeletal dysplasia caused by a Lys650Met mutation in fibroblast growth factor receptor 3. American journal of medical genetics. 1999 Jul 2:85(1):53-65 [PubMed PMID: 10377013]
Level 3 (low-level) evidenceSmid CJ, Modaff P, Alade A, Legare JM, Pauli RM. Acanthosis nigricans in achondroplasia. American journal of medical genetics. Part A. 2018 Dec:176(12):2630-2636. doi: 10.1002/ajmg.a.40506. Epub 2018 Oct 31 [PubMed PMID: 30380187]
Dai L, Xie L, Wang Y, Mao M, Li N, Zhu J, Kim C, Zhang Y. A novel COMP mutation in a pseudoachondroplasia family of Chinese origin. BMC medical genetics. 2011 May 21:12():72. doi: 10.1186/1471-2350-12-72. Epub 2011 May 21 [PubMed PMID: 21599986]
Level 3 (low-level) evidenceOzcetin M, Arslan MT, Karapinar B. An achondroplasic case with foramen magnum stenosis, hydrocephaly, cortical atrophy, respiratory failure and sympathetic dysfunction. Iranian journal of pediatrics. 2012 Mar:22(1):121-4 [PubMed PMID: 23056871]
Level 3 (low-level) evidence