
 Calcium Deposition and Other Renal Crystal Diseases
Calcium Deposition and Other Renal Crystal Diseases
Introduction
Crystals are groups of ions, atoms, and molecules that can form into homogeneous solids and have a fixed distance between the constituent parts, creating an orderly, repeating 3-dimensional pattern. The molecular components of crystals are filtered from the plasma through the kidneys and concentrated in the renal tubules. The kidneys are a primary site of crystalline deposition, receiving 25% of total cardiac output.
These deposits primarily occur in the renal tubules, interstitium, or both. They are associated with the development of acute kidney injury (AKI) or progressive chronic kidney disease (CKD), which can present with microscopic hematuria, crystalluria, and sub-nephrotic range proteinuria.[1][2][3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Urinary and renal crystalline disorders are broadly categorized by underlying etiology into four major groups.[3][4]
- Calcium oxalate and calcium phosphate crystallopathy
- Paraproteinemia-related crystal deposition diseases
- Crystal deposition secondary to metabolic and inherited disorders
- Drug-induced crystal deposition disease
Epidemiology
Autosomal recessive crystalline deposition diseases include the following: primary hyperoxaluria; 2,8 dihydroxy adenine (2,8 DHA) crystalluria; cystinuria, and cystinosis. These diseases are more prevalent in areas with consanguineous marriages.
Primary hyperoxaluria type 1 (PH1) accounts for 1% to 2% of end-stage renal disease (ESRD) in children. PH1 has an incidence of 1 in 120,000 live births in Europe.
Cystinosis occurs approximately once in every 100,000 to 200,000 live births.[5] It accounts for 5% of ESRD in children.
Cystinuria accounts for about 1% to 2% of renal stones in adults.[6]
Adenine phosphoribosyl transferase deficiency (APRT), which is the cause of 2,8 DHA crystalline nephropathy, has an estimated prevalence of about 400 worldwide. While very rare, most cases are found in France, Iceland, and Japan.[7]
Multiple myeloma is diagnosed in an estimated 34,920 people in the US and approximately 588,161 people worldwide each year.
Pathophysiology
Crystal deposition leads to tubular obstruction, inflammation, and intracellular inclusion. As the crystals are phagocytosed by lysosomes of the renal tubular cells, they release cathepsin-B and other cell death pathway mediators, leading to eventual necroptosis of the cell.
Renal tubular cell necrosis stimulates the inflammatory pathway leading to further cell damage, activation of Toll-like receptors, induction of NLRP3 inflammasome activity, and secretion of interleukin 1b, which further leads to increased transcription and expression of several inflammatory cytokines.
Activation of the inflammatory cascade contributes to the robust inflammation within the tubulointerstitium, which plays a key role in the development of acute and chronic kidney disease in renal crystallopathies.[2][3]
Hyperoxaluria and calcium oxalate crystal deposition in the vagina has been proposed as possible etiology of vulvodynia in women. While theoretically possible, the scientific data does not support this conclusion except in rare, anecdotal cases.[10][11][12][13][14]
Histopathology
Calcium oxalate crystals are deposited in the renal tubules, interstitium, or both, leading to AKI, oxalate nephropathy, and progressive CKD.
The prevalence of oxalate nephropathy varies across different studies. The prevalence was about 1% on native kidney biopsies in one Belgian series but reached up to 4% in a biopsy series done in the New York metro area.[2][4][15]
Oxalate is the ionized form of oxalic acid. The average person consumes about 100 mg to 140 mg of oxalate daily. Only 5% to 15% of dietary oxalate is absorbed in the intestinal tract. Most dietary oxalate binds to ingested calcium and is excreted harmlessly as calcium oxalate in the stool. Oxalate is also degraded by colonic bacteria such as Oxalobacter formigenes. The liver produces 60% to 80% of plasma oxalate, and dietary absorption contributes to the rest.[16]
Oxalate has no physiological role in the human body. Most of the absorbed oxalate gets filtered and excreted unchanged by the glomerulus and is actively secreted by the proximal convoluted tubules into the urine.[17]
The urinary oxalate excretion varies depending on daily oral intake; oxalate excretion exceeding 40 mg per 24 hours is generally consistent with hyperoxaluria. Hyperoxaluria is an independent risk factor for the progression of chronic kidney disease, even in patients with no evidence of oxalate nephropathy.[17]
Systemic levels of oxalate increase once the GFR drops below 30 ml/min, which leads to extrarenal calcium oxalate deposits in other organs, such as the bones, skin, central nervous system, retina, and cardiovascular system, consistent with systemic oxalosis.
Calcium oxalate crystals are translucent when viewed by standard light microscopy and strongly birefringent on polarized microscopy.
There are two crystalline forms of calcium oxalate. Calcium oxalate monohydrate can appear dumbbell-shaped. Calcium oxalate dihydrate looks like two four-sided pyramids connected at the bases and is also sometimes described as envelope-shaped. Calcium oxalate monohydrate stones are tough and difficult to break down, while calcium oxalate dihydrate calculi are quite fragile. Many stones and crystals will have varying percentages of each.
There are three categories of hyperoxaluria: primary (genetic), secondary (acquired), and enteric.[16]
Primary hyperoxaluria is a group of rare autosomal recessive disorders associated with excess oxalate production by the liver. The high oxalate load exceeds the excretion capacity of the kidneys and causes precipitation, leading to recurrent kidney stones and oxalate nephropathy. Patients with primary hyperoxaluria have abnormally elevated urinary oxalate excretion, typically in the range of greater than 100 mg/day, but a threshold of greater than 75 mg/day is generally used to differentiate primary and secondary hyperoxalurias. For more information on primary and secondary hyperoxaluria, please see our companion StatPearls reference article on "Hyperoxaluria."[16]
Primary hyperoxaluria is a common cause of ESRD in younger patients. It has three subgroups based on the specific type of enzymatic defect.[16]
- Type 1 Primary Hyperoxaluria (PH1) is the most common and severe type of primary hyperoxaluria.
- The deficiency of pyridoxal-dependent, alanine glyoxal transferase in hepatic peroxisomes leads to increased oxalate production from glyoxylate in the cytoplasm of liver cells.
- The AGXT gene encodes AGT, and more than 200 mutations of the AGXT gene are known to cause PH1.[1][2][17]
- Patients with PH1 have recurrent calcium oxalate kidney stones, rapidly progressive chronic kidney disease, and can progress to ESRD.
- The median onset of ESRD is 5.5 years, and most develop renal failure in the first three decades of life; some patients have delayed presentation in adulthood with occasional or recurrent calcium oxalate kidney stones as their only clinical manifestation.[2]
- Primary hyperoxaluria should be considered in children with kidney stones or nephrocalcinosis and adults with recurrent calcium oxalate kidney stones.[17]
- Type 2 Primary Hyperoxaluria (PH2) is caused by a glyoxylate hydroxypyruvate reductase (GRHPR) deficiency, leading to excess glyoxylate accumulation, an immediate oxalate precursor.
- This enzyme is expressed in multiple other tissues in addition to the liver.[16]
- Patients with PH2 disease can have progressive CKD and recurrent calcium oxalate nephrolithiasis, and recent data suggest that these can also progress to ESRD, requiring renal transplantation.
- Type 3 Primary Hyperoxaluria (PH3) is caused by the deficiency of 4-hydroxy-2-oxoglutarate (HOGA).
Secondary hyperoxaluria causing oxalate nephropathy is characterized by increased oxalate levels due to various secondary causes that include increased dietary intake of oxalate-containing foods, increased intestinal absorption of oxalate in a setting of fat malabsorption (enteric hyperoxaluria), increased intake of oxalate precursors, and alteration of intestinal microbial flora.
The prevalence of this condition varies from 1% to 4% on native kidney biopsies. Patients with oxalate nephropathy showed progressive advanced CKD (after other etiologies of renal failure were excluded) and had deposition of calcium oxalate crystals within the tubular epithelial cells, lumens, and occasionally in the interstitium; most of these patients had a hyperoxaluria-enabling condition.[4]
Excess intake of oxalate precursor substances like ethylene glycol and vitamin C is known to cause crystalline oxalate nephropathy. Cases of vitamin C-induced oxalate nephropathy are associated with mega doses of vitamin C intake, such as those seen during the COVID-19 pandemic. Cases can also occur with the intake of other oxalate precursors such as methoxyflurane (a general anesthetic), ethylene glycol, and naftidrofuryl oxalate (a vasodilator used in the treatment of peripheral vascular disease).[1][2]
Excess intake of oxalate-containing foods such as nuts (peanuts, cashews, and almonds), spinach, rhubarb, star fruit, chaga mushroom powder, and black iced tea causing oxalate nephropathy have been reported in multiple case reports.[1][16] Case reports involving acute ingestions causing oxalate nephropathy often show rapidly improved renal function, suggesting a better prognosis than other causes of oxalate nephropathy.
Alteration of intestinal microbial flora is also a potential cause of hyperoxaluria. Commensal gut bacteria such as Oxalobacter formigenes and other bacteria can metabolize intestinal oxalate, thus preventing absorption. Antibiotics, such as fluoroquinolones that kill these normal intestinal bacteria, can be associated with an increased risk for oxalate stones and nephropathy. Patients with diabetes and obesity can also have elevated gut oxalate absorption and hyperoxaluria through unclear mechanisms.[16]
Patients with oxalate nephropathy commonly have precipitating events such as acute dehydration, diuretic use, inflammation, recent antibiotic use, or renin-angiotensin-aldosterone system (RAAS) inhibitors, suggesting a combination of a hyperoxaluria-enabling condition with a precipitating event leading to oxalate deposition causing kidney damage.[16]
Enteric hyperoxaluria occurs in a setting of intestinal bypass surgery or short-bowel syndrome, resulting in calcium-sequestering steatorrhea due to reduced free intestinal calcium, causing increased gastrointestinal oxalate absorption and severe hyperoxaluria.
Usually, dietary oxalate binds to dietary calcium, forming insoluble calcium oxalate, which is excreted in the stool. Conditions that cause fatty acid diarrhea lead to increased binding of fatty acids to dietary calcium, making it unavailable to bind to dietary oxalate. Unbound soluble dietary oxalate gets absorbed into the systemic circulation, increasing oxalate excretion in urine. An intact colon appears to be required for gastrointestinal oxalate absorption.
Oxalate nephropathy and nephrolithiasis were seen in as many as 35% of patients after jejunoileal bypass, associated with increased morbidity, which contributed to the abandonment of that procedure in 1979. It was also seen after the Roux-en-Y gastric bypass procedure. It does not occur with restrictive weight loss surgeries like gastric sleeve procedures, as the small bowel is not bypassed.[18]
- Oxalate nephropathy can be seen in chronic pancreatitis, celiac disease, Crohn disease, cystic fibrosis, and with the use of somatostatin analogs.
- Drugs like orlistat can cause oxalate nephropathy by impairing fatty acid absorption.
- Moderate to severe hypocalcemia can be seen in patients with chronic pancreatitis with oxalate nephropathy, which can be a clue to the diagnosis.[2][4]
Calcium phosphate deposition disease is caused by the development of calcium phosphate crystals within the renal tubules and interstitium, leading to crystalline nephropathy. Calcium phosphate deposition should be especially considered in patients with nephrocalcinosis.
Differentiation can be seen in microscopic analysis. Calcium phosphate crystals stain blue or red on hematoxylin and eosin (H&E) stain, whereas calcium oxalate crystals are translucent. The von Kossa stain is specific for phosphate, so it only stains calcium phosphate, not calcium oxalate. Another differentiating feature is that calcium oxalate crystals are birefringent on polarized microscopy.[1][2][4]
Conditions causing hypercalcemia, hypercalciuria, hyperphosphatemia, hyperparathyroidism, hyperphosphaturia, and persistently alkaline urine are the common causes of calcium phosphate deposition.
Exposure to bowel preparations containing high amounts of phosphate was previously a common cause of calcium phosphate nephropathy, prompting an FDA warning regarding the risk for phosphate nephropathy with sodium phosphate bowel preparations in December 2008, which ultimately led to the voluntary withdrawal of over-the-counter sodium phosphate bowel preparations.[4][19]
Crystal Deposition Secondary to Metabolic and Inherited Disorders
2,8-Dihydroxyadenine (2,8 DHA) crystallopathy is an autosomal recessive disorder secondary to the adenine phosphoribosyl transferase enzyme deficiency leading to crystalluria, CKD, and ESRD. The adenine phosphoribosyl transferase enzyme converts the adenine to adenine-phosphate: the lack of this enzyme leads to increased conversion of adenine to 8-hydroxy adenine and 2,8-dihydroxy adenine production through the xanthine oxidase pathway.
2,8-dihydroxy adenine crystals are highly insoluble in urine, leading to tubular precipitation, crystalluria, recurrent nephrolithiasis, progressive kidney disease, and ESRD.[3][4] 2,8 DHA crystals are radiolucent and can easily be incorrectly diagnosed as uric acid stones if not chemically analyzed. 2,8 DHA is also a rare type of crystalline nephropathy, the diagnosis of which is often missed before renal transplantation. Many of these patients are unfortunately only diagnosed with this disease after they develop evidence of posttransplant allograft dysfunction.
These crystals are birefringent, reddish-brown, and have a Maltese cross appearance on polarized microscopy. Renal biopsy specimens show intratubular crystals, which are irregular and fan-shaped. They stain brownish green on H&E and periodic acid-Schiff, light blue on Trichrome, and positive for silver staining. Pathologists need to differentiate these crystals from calcium oxalate crystals as they have a similar microscopic appearance.
Patients can develop renal calculi, acute renal injury, and progressive CKD with a delay in diagnosis. This disease has no extrarenal manifestations as these crystals are deposited only in the kidneys. A recent observational study suggests that many patients can have progressive CKD without a history of kidney stones.[3][4][7][20]
Dent disease and Lowe syndrome are a group of X-linked recessive disorders secondary to mutations in CLCN5 and OCRL1 genes leading to Dent disease type 1 and type 2, respectively. A certain percentage have no identifiable gene mutation and are classified as Dent disease 3. X-linked recessive disorders, such as Dent disease and Lowe syndrome, are also associated with hypercalciuria and renal calcium phosphate deposition, causing nephrocalcinosis, progressive renal failure, and, ultimately, ESRD.[3][4]
- Patients can present with proximal tubulopathy, full-blown Fanconi syndrome, or hypercalciuria. The cause of the hypercalciuria is secondary to a combination of increased calcium absorption and defective proximal tubular calcium uptake. Clinical presentation can be variable, even within the same family.
- The initial presentation may include calcium oxalate and calcium phosphate crystalline renal disease (nephrocalcinosis) and can progress to chronic kidney failure and ESRD. Such individuals often have renal phosphate wasting, leading to osteomalacia. Those patients with Dent disease type 2 can present with other signs like hypotonia, cataracts, and intellectual disability.[3][4]
Lowe syndrome is another X-linked recessive disorder secondary to a mutation in the OCRL1 gene, similar to Dent disease type 2. Patients develop proximal tubulopathy, hypercalciuria, phosphaturia, nephrocalcinosis, osteomalacia, progressive renal failure, metabolic acidosis, and ESRD. Patients usually have associated signs such as cataracts, neurologic signs, developmental disability, and hypotonia. These patients tend to have rapid progression of renal failure and rapid development of ESRD.[3][4]
Treatment is primarily supportive for both diseases, including thiazide diuretic therapy to reduce urinary calcium excretion, oral potassium citrate, phosphate supplementation, and vitamin D repletion.
Cystinosis is a rare autosomal recessive disorder characterized by lysosomal accumulation of cystine, secondary to defective transport, leading to intracellular crystallization. The disease is caused by a mutation in the CTNS gene, which encodes cystinosin, the lysosomal cystine transporter which moves cystine extracellularly.[21][22]
The lack of a functional transporter causes accumulation and crystallization of the cystine within the lysosome, which ultimately causes apoptosis and localized tissue damage.[23] This disease can manifest in infancy as failure to thrive. A few cases have been shown to occur in adolescence or adulthood. Both infantile and adolescent presentations can occur in different members of the same family.[3][4]
Some patients can develop proximal tubular dysfunction leading to a full-blown Fanconi syndrome, and progressive renal failure and ESRD can occur. It is the most common cause of Fanconi syndrome in children, and screening of newborns is performed in most developed countries. Early initiation with the cystine-depleting—therapy cysteamine is key to delaying CKD progression.[24]
Pathology shows cystine crystal accumulation in glomerular podocytes, mesangial cells, interstitial macrophages, and occasionally in tubular cells. Crystals are needle-like, have geometric shapes, and are strongly birefringent on polarized microscopy; these crystals are typically dissolved during processing. The finding of multinucleated podocytes is often helpful in diagnosis.[25][26][27] New biomarkers for this condition are currently being studied.[28]
Cystinuria causes urinary cystine stones with characteristic hexagonal-shaped cystine crystals in the urine. Cystine stones can be difficult to see on X-ray as they are relatively radiolucent. They are very hard and relatively resistant to treatment. Cystine urinary crystals are increasingly soluble as the urinary pH increases, so treatment includes raising the urine pH to 7.5 or even 8 using potassium citrate and sodium bicarbonate.[29]
Very high hydration levels are needed to generate the recommended 3 liters of urine production every 24 hours. When simple measures like hydration and alkalinization cannot control cystine crystallization and stone formation, tiopronin is used as a urinary cystine binder, making it far more soluble.[29] For more information on cystine, cystine stones, and cystinuria, see our companion Statpearls reference article on "Cystinuria."[29]
Uric acid crystal renal deposition can cause acute kidney injury, CKD, or nephrolithiasis with obstructive uropathy. Acute urate nephropathy typically presents as oliguric or anuric renal failure, commonly seen in conditions of massive cell destruction such as tumor lysis. Uric acid crystals associated with acute kidney injury are predominantly seen in the renal collecting tubules.
Uric acid crystals are dissolvable in formalin-fixed tissue, leading to empty lacunae. Alcohol fixed or frozen section specimens show needle-shaped or rectangular crystals that stain blue with H&E and are birefringent on polarized microscopy.[30][31]
Treatment primarily consists of aggressive hydration, urinary alkalinization, and using xanthine oxidase inhibitors or recombinant urate oxidase, which converts the urate to water-soluble allantoin. While both treatment options can be used prophylactically, recombinant urate oxidase is particularly useful even after the development of acute urate nephropathy. Chronic urate nephropathy is reviewed elsewhere.[3][4][30][31] For more information on uric acid urolithiasis, urinary uric acid, and hyperuricemia, see our companion Statpearls reference articles on "Uric Acid Nephrolithiasis," Hyperuricosuria," and "Hyperuricemia."[32][33][34]
Paraproteinemia-related Crystal Deposition Diseases
Dysproteinemia is characterized by the clonal proliferation of immunoglobulins (Igs) or Ig subunits associated with plasma cell neoplastic or B-cell lymphoproliferative disorders. Diagnosis is generally by renal biopsy findings, serum protein electrophoresis, and skin histological examinations.
Paraproteinemia-related crystal deposition disease is associated with five major types of renal crystallopathies, which are summarized below.
1. Light Chain Proximal Tubulopathy
- Glomeruli freely filter free light chains into the tubular lumen, and the filtered light chains are endocytosed by the proximal tubular cells through megalin and cubilin receptors, which become overwhelmed by large amounts of light chains produced in paraproteinemia; this leads to intracellular crystallization in proximal tubular cells and light chain proximal tubulopathy.[35]
- Light chain proximal tubulopathy is characterized by the accumulation and deposition of light chain monoclonal Igs in the proximal tubular cells, leading to proximal tubular injury and dysfunction. This can cause Fanconi syndrome, characterized by glucosuria, aminoaciduria, uricosuria, and phosphaturia. The proximal tubular changes can be subtle, and some patients may not develop any clinical signs suggesting proximal tubular dysfunction.[36][37]
- The crystals formed are needle-shaped and weakly birefringent on polarized microscopy. With staining, they appear eosinophilic, red on mason trichrome, and typically pale with PAS. The standard immunofluorescence techniques can often miss these crystals on frozen section specimens as they can be inaccessible to antibody binding.
- Improved sensitivity is obtained on formalin-fixed immunofluorescence stains, antigen retrieval techniques, or protease enzyme use. An electron microscopic examination of the proximal tubular cell reveals intracytoplasmic inclusion crystals.[3][4]
- Light chain Fanconi syndrome is commonly associated with smoldering myeloma and monoclonal gammopathy of undermined significance (MGUS); rare cases have also been reported with chronic lymphocytic leukemia and diffuse large B-cell lymphoma.[38]
- All crystals causing light chain Fanconi syndrome belong to the K variant light chain, derived from the VK1 sub-group.[3][4]
- Treatment of the underlying clonal proliferative disorder is recommended to treat light chain Fanconi syndrome, while plasma exchange has no role in its therapy.[39][40]
- Other recommended treatments include bortezomib, autologous stem cell transplantation, immunotherapy drugs, daratumumab, and renal transplantation.[39]
2. Light Chain Cast Nephropathy
- Light-chain cast nephropathy, also called myeloma cast nephropathy, is the most common variety of paraproteinemia-related renal disorders.
- It is characterized by monoclonal light chains admixed with Tamm-Horsfall protein in the thick ascending loop of Henle and deposited in the distal convoluted tubule.
- Excess light chains enter the distal convoluted tubules when the proximal tubular cells are supersaturated and interact with Tamm-Horsfall protein (THP).
- Using loop diuretics and the presence of hypercalcemia increases the risk for light chain cast nephropathy by facilitating the interaction between the light chains and the THP.
- ACE inhibitor, ARB, and NSAID use also increase the risk of cast nephropathy.[41]
- Leukocyte activation by the THP contributes to the interstitial inflammation and fibrosis seen in myeloma cast nephropathy.[42]
- Increased activation of the NF-kB pathway and mitogen-activated protein kinases cause proximal tubular cell injury and necrosis, which leads to further production of inflammatory mediators such as IL-6, IL-8, and TNF-β1.
- Increased inflammatory response plays a crucial role in tubular interstitial atrophy and fibrosis.[42]
- The casts are hypereosinophilic, pale with PAS staining, polychromatic, lamellated with mason trichrome stain, and weakly birefringent on polarized microscopy. They have a hard texture, often fracture when cut with a microtome (leading to sharp or jagged edges), and can have a fractured appearance. On electron microscopy, these casts exhibit sharp edges, geometric shapes, and cellular reactions by tubular epithelial cells, infiltrating leukocytes, and giant cells.[3][4]
- Immunofluorescence plays a critical role in diagnosing light chain cast nephropathy; staining is positive for either Kappa or Lambda chains, with minimal staining of the opposite light chain.[36]
- Treatment is aimed at stopping the production of monoclonal immunoglobulins and is similar to the management of light chain proximal tubulopathy described above.
- Rapid lowering of free light chains with chemotherapy has been associated with renal functional recovery and is considered renoprotective.[43]
- Plasma exchange may be of some benefit in this disorder but is controversial and, if used, should be combined with chemotherapy.[44][45]
- The beneficial role of plasmapheresis has not been proven in randomized trials and, therefore, is not currently recommended for the treatment of light chain cast nephropathy.[43]
3. Crystalglobulinemia
- This rare disorder is associated with paraproteinemia and multiple myeloma, as well as other malignancies, and is characterized by the accumulation of hypereosinophilic extracellular microcrystals of monoclonal proteins primarily within the systemic and renal microvasculature. These microcrystals can cause vascular injury with thrombosis and occlusion, leading to end-organ damage.[3][46]
- Patients typically present with acute kidney injury, renal failure, albuminuria, arthralgias, and rashes.[46]
- On histological examination, eosinophilic crystal deposits are seen within the microvasculature, renal glomeruli, and occasionally in the renal tubules.[46] The crystals have geometric shapes, stain red on trichrome, and pale on PAS. Proliferative glomeruli may show engulfed crystals within the leukocytes and monocytes. IgG Kappa- or Lambda-restricted staining is noted on immunofluorescence.[46]
- Treatment is aimed at reducing paraprotein production. Plasma exchange is recommended for the rapid removal of these circulating paraproteins.
- Data is limited on this rare disease, but the prognosis is generally poor, especially if renal failure is the initial presenting symptom.[3][46]
4. Crystal-storing Histiocytosis
- Crystal-storing histiocytosis is an extremely rare entity associated with B-cell malignancies, such as multiple myeloma, characterized by needed-shaped crystal accumulation of monoclonal immunoglobulins in the histiocytes.[47] These histiocytes are typically found in the bone marrow, and they can also be occasionally seen in extranodal tissues, the lungs, lymph nodes, skin, stomach, eyes, and kidneys.[48]
- This may present about 25% of the time as a localized mass, swelling, or lesion incidentally found on imaging or physical examination that may initially be considered suspicious for cancer. It can be easily confused with an organ malignancy such as lung cancer.[47]
- This entity is the same as monoclonal proximal tubulopathy except that the crystals accumulate in histiocytes rather than in proximal tubular cells. All these crystals also belong to the Kappa variant, derived from the VK1 sub-group.[48]
- The differential diagnosis includes malakoplakia, Gaucher disease, and rhabdomyoma.[47]
- Treatment should be aimed at stopping paraprotein production, and plasma exchange is not recommended.[3][4]
There are a few case reports of patients having light chain Fanconi syndrome, light chain cast nephropathy, and crystal-storing histiocytosis together. All these patients had a monoclonal kappa neoplasm derived from the VK1 sub-variant.[37][48]
5. Light Chain Crystalline Podocytopathy
- This is also a very rare entity described primarily in case reports and can be associated with other paraprotein-related renal crystalline diseases, such as crystal-storing histiocytosis or light chain proximal tubulopathy.[49]
- It has been associated with focal and segmental glomerulosclerosis (FSGS), which is thought to be secondary FSGS from podocyte injury stemming from crystal deposition.[50]
- Clinical manifestations are related to proteinuria and possible nephrotic syndrome. Monoclonal IgG-κ is the most commonly involved paraprotein.[50][51]
- Similar to other renal crystallopathies, immunofluorescence of the light chains may be negative due to the epitope binding site not being exposed. Staining is improved with antigen exposure techniques.[50][49][50]
Drug-induced Renal Crystalline Deposition Diseases
Various medications used in clinical practice can be associated with crystal deposition within the renal tubules and parenchyma, causing renal tubular crystallopathy and acute kidney injury. Drug-induced crystal deposition diseases commonly occur in the setting of volume depletion, use of higher than recommended dosages, prolonged medication use, and changes in urine pH. All of these can contribute to supersaturation and drug or metabolite precipitation. The pK value of the specific drug is important in determining its crystallization potential.
Clinicians should consider the potential risk for crystalluria and acute kidney injury with these drugs. A thorough clinical history and examination of the urine sediment are helpful and critical in the diagnosis.
Acyclovir
- The higher dose of acyclovir used by the IV route (rather than by mouth) and rapid infusion can cause crystalluria and acute kidney injury.
- Acyclovir crystals are needle-shaped, birefringent on polarized microscopy, and seen in the renal distal tubules.
- Avoiding rapid infusions of IV acyclovir at higher doses and maintaining adequate hydration helps prevent crystalluria.[3][4][26]
Amoxicillin
- Amoxicillin is a commonly used penicillin group antibiotic; it can be associated with crystalline nephropathy. Risk factors for crystallization include a higher dose of the medication, volume depletion, and an acid urine pH.
- The crystals are needle or rod-shaped and can form bunches or sheaves. The treatment involves stopping the medication, volume repletion, and urinary alkalization when the urine pH is lower than 6.
- No histological evidence of intrarenal deposits of amoxicillin crystals has been published despite their known association with crystalluria and AKI, like sulfadiazine and sulfamethoxazole.[3][4][26]
Ciprofloxacin
- Ciprofloxacin, a fluoroquinolone antibiotic, can be associated with crystalluria in an alkaline urinary pH setting and with higher antibiotic doses.
- Ciprofloxacin crystals can assume different shapes and can appear as needles, starbursts, sheaves, tans, and butterflies.
- Crystals have a lamellar appearance, and they are strongly birefringent on polarized microscopy.
- Crystalline nephropathy can be prevented by volume repletion, dose adjustment according to renal function, and prevention of urinary alkalization.[3][4][26]
Methotrexate
- Methotrexate can cause renal damage and AKI by various mechanisms, but crystal deposition is the most common cause of acute kidney injury, usually reversible.[3][4][26]
- The risk for crystal precipitation is higher in conditions associated with volume depletion, higher doses, and low urinary pH.[3][26]
- Methotrexate crystals form annular structures of needle-shaped crystals that stain yellow, golden, or brown on an H&E stain. They are strongly birefringent on polarized microscopy.
- Intratubular precipitation is prevented by maintaining high urinary flow and urinary alkalization with sodium bicarbonate or potassium citrate, optimally with a pH greater than 7.[3][4][26]
Protease Inhibitors (indinavir, atazanavir, and darunavir)
- All protease inhibitors have been associated with crystalluria and crystalline nephropathy. These drugs precipitate in urine with a pH higher than 3.5.
- Nephrotoxicity is prevented by discontinuing the drug and maintaining adequate hydration to promote a euvolemic state. Urinary acidification is not recommended to treat crystalluria.
- These drugs can also cause radiolucent stones, which are not visible on non-contrast CT scans or X-rays. Patients taking one of these medications who present with symptoms of renal colic and hydronephrosis require a contrast study to help identify potential medication stones.[52][53]
- Indinavir is the most studied in this class. Its crystals are translucent, needle-shaped, typically clustered in distal tubules and collecting ducts, associated with monocytic inflammatory infiltration and giant cell reaction. Darunavir crystals are needle-shaped or biconvex and are birefringent on polarized microscopy.[3]
- Early recognition of the disease can avoid irreversible renal fibrosis and the development of CKD.[3][4][26]
Sulfa Antibiotics
- Sulfadiazine is commonly associated with crystalluria, but sulfamethoxazole can also cause urinary crystals, although less commonly.
- A higher incidence of sulfadiazine-associated crystalluria has been noted with the increased use of pyrimethamine, a sulfa medication used for toxoplasmosis.[54][55]
- Sulfa crystals have hourglass shapes with prominent radial striations and can have a "sheaves of wheat" appearance. They are birefringent on polarized microscopy and can be deposited in any part of the renal tubules or urinary tract.[26]
- IV hydration and alkalinization of the urine promote the solubility of these crystals and renal recovery.
- Sulfonamide-induced crystalluria and AKI are considered reversible. A renal biopsy may be necessary if there is no improvement in renal function, as these drugs can also cause acute interstitial nephritis.[4][26]
Triamterene
- Triamterene is a potassium-sparing diuretic commonly used in combination with thiazide diuretics. Crystalluria is common with triamterene, but AKI is rare.
- Triamterene precipitates in acidic urine.
- Its urinary crystals are brown, can form Maltese cross-like structures, and are birefringent on polarized microscopy.
- It is essential to differentiate these crystals from 2,8 dihydroxyadenine crystals as they appear similar in urine and renal pathology specimens.[3][4][26]
- Amiloride is an alternative potassium-sparing diuretic that can be substituted for triamterene and is not associated with crystalluria or urolithiasis.
History and Physical
A thorough history, physical examination, complete family history, examination of dietary habits, review of medications, and supplement intake are essential steps when evaluating patients with suspected renal crystallopathy.
Patients with primary hyperoxaluria can have recurrent episodes of kidney stones causing abdominal pain, renal colic, hematuria, dysuria, urinary tract infections, or, in children, failure to thrive.[56]
- Primary hyperoxaluria should be considered in children with kidney stones or nephrocalcinosis and adults with recurrent calcium oxalate kidney stones along with markedly elevated 24-hour urine oxalate excretion, typically greater than 75 mg.[16][17]
- Patients with primary hyperoxaluria will have recurrent calcium oxalate kidney stones, rapidly progressive CKD, and usually progress to ESRD.
- The median onset of ESRD is 5.5 years, and most develop renal failure in the first three decades of life. However, some can have a delayed presentation in adulthood with occasional or recurrent calcium oxalate kidney stones as the only clinical manifestation.
- Patients with primary hyperoxaluria can have deposition of calcium oxalate crystals in the macula and hyperpigmentation of the retina with or without retinal edema.
- Some patients can develop oxalate deposition in bones, leading to skeletal malformations and multiple fractures. The presence of a sclerotic band in rapid-growth areas is an early radiological sign of bone oxalosis.
- Oxalate crystal precipitation in the bone marrow can cause inflammation and fibrosis, leading to stimulation of extramedullary hematopoiesis. These patients can develop pancytopenia, hepatomegaly, and splenomegaly.
- Cardiac deposition of calcium oxalate crystals can cause cardiac arrhythmia, heart block, and cardiomyopathy.
- Dermal deposition of oxalate crystals can cause subcutaneous nodules, eschar formation, and superficial ulceration.
- Cardiac and dermal manifestations are uncommon in infants, but these can be found in children and adults.
- Some patients can have neuropathy, hypogonadism, and hypothyroidism.
- Some patients with primary hyperoxaluria can have vascular deposition of calcium oxalate. Patients with crystaloglobulinemia may have vascular deposition of paraprotein crystals, causing livedo reticularis, acrocyanosis, peripheral gangrene, and ulcerations.
Individuals with secondary hyperoxaluria and 2,8 DHA crystalline nephropathy can have a history of recurrent nephrolithiasis, progressive chronic kidney disease, and nephrocalcinosis. Patients with PH2 disease can also have progressive CKD and recurrent calcium oxalate nephrolithiasis, and recent data suggest that these can progress to ESRD, requiring renal transplantation.
Progression to CKD/ESRD has yet to be documented with PH3.[1][2][17]
Patients with cystinosis will accumulate cystine crystals in the renal tubules, contributing to progressive renal dysfunction and ESRD. They can also develop accumulations of cystine in the eyes, bones, thyroid gland, pancreas, gonadal organs, liver, muscles, and brain.
- Cystine accumulates predominantly in the kidneys and the eyes, primarily found in the cornea, posterior chamber, and retina.
- Cystine accumulation in the thyroid gland can cause atrophic thyroiditis, leading to hypothyroidism; pancreatic deposition of cystine crystals can cause pancreatic insufficiency and diabetes; and muscle accumulation can cause myopathy. Patients with significant myopathy would be at risk for developing aspiration pneumonia and extrapulmonary restrictive lung disease.
- Some patients can have cerebral accumulations causing multiple central nervous system symptoms, bone accumulation leading to growth retardation, and multiple fractures.[5]
Patients with paraproteinemia can present with fatigue, bone pain, anemia, renal dysfunction, hypercalcemia, increased total serum protein level, lytic bone lesions, and compression/pathological fractures.
Patients with paraproteinemia-related Fanconi syndrome, cystinosis, Dent disease types 1 and 2, and Lowe syndrome usually demonstrate Fanconi syndrome. They may also present with nephrocalcinosis and hypercalciuria.
Patients with proximal tubular dysfunction can have episodes of dehydration, volume depletion, metabolic acidosis, hypophosphatemia, and AKI.
Infants and children with proximal tubular dysfunction can develop failure to thrive, growth retardation, osteomalacia, and bone fractures.
Patients with crystaloglobulinemia can have cutaneous purpura, peripheral ulcers, mucosal ulcers, erosive polyarthropathy, peripheral neuropathy, peripheral ischemia, renal dysfunction, and rare visceral ischemia.
Evaluation
Patients with suspected primary hyperoxaluria should receive a 24-hour urine excretion for calcium, phosphorus, magnesium, uric acid, oxalate, citrate, and cystine. Any renal stones should be collected and chemically analyzed.
Hyperoxaluric patients should have an evaluation to rule out secondary causes of hyperoxaluria. Those with severe hyperoxaluria (>75 mg/day) should undergo genetic testing to confirm the diagnosis of primary hyperoxaluria. There are also reported cases of hypercalcemia in patients with primary hyperoxaluria.[56][57]
Plasma oxalate levels should be measured in patients with a decline in renal function, especially when the GFR is below 30 mL/min, as the urinary oxalate levels will not be representative. In primary hyperoxaluria patients with ESRD, plasma oxalate levels are typically higher than 80 μmol/L, while in non-primary hyperoxaluria hyperoxaluric patients, the plasma oxalate level may range between 30 μmol/L to 80 μmol/L.[58][59]
Spot urine tests for glycolate, glycerate, and 4-hydroxy-2-oxoglutarate have a high sensitivity, particularly for PH2 and PH3.[60]
Genetic testing is the gold standard for diagnosing all types of primary hyperoxaluria; it should be performed in all patients with suspected primary hyperoxaluria (a 24-hour urine oxalate >75 mg in patients without enteric hyperoxaluria), and it also helps estimate the responsiveness to pyridoxine treatment in PH1 patients.[16][17]
Systemic levels of oxalate increase once the GFR drops below 30 mL/min, which leads to extrarenal calcium oxalate deposits in other organs, such as the bones, skin, central nervous system, retina, and cardiovascular organs, consistent with systemic oxalosis.[17] These patients should get an electrocardiogram, echocardiogram, thyroid function panel, and x-ray examination of the long bones to rule out fractures and radiodense metaphyseal bands.[61]
- Patients suspected of Fanconi syndrome should undergo microscopic urinalysis and an evaluation for glycosuria in the presence of normal serum glucose levels, aminoaciduria, uricosuria, and phosphaturia. Serum chemistry, calcium, phosphorous, and uric acid levels should be obtained. See our companion StatPearls reference article on "Fanconi syndrome."[62]
- Cystinosis is diagnosed by the detection of elevated cystine levels in leukocytes.
- It can also be diagnosed clinically by detecting corneal cystine crystals on slit lamp examination.
- Molecular testing is another well-established test to diagnose cystinosis.
- Plasma chitotriosidase enzymatic activity is an excellent predictor of leukocyte cystine levels.
- 2,8 DHA crystalline nephropathy, Dent disease types 1 and 2, and Lowe's syndrome can be diagnosed and confirmed by genetic testing. However, many patients with these conditions do not have detectable mutations, so this is not an exclusionary test.
- Patients with suspected paraproteinemia should undergo a complete blood count with differential, a comprehensive metabolic panel including calcium level, serum protein electrophoresis with immunofixation, serum free light chain testing, 24-hour urine for electrophoresis with immunofixation, serum beta -2 microglobulin level, albumin, and LDH. They should also undergo imaging of the entire skeleton, preferably whole-body, low-dose computed tomography, as a whole-body skeletal survey is less sensitive.[9]
- Those with an abnormally elevated serum-free light chain immunoglobulin level greater than 500 milligrams/liter should have proteinuria quantification.
- Patients with predominantly albuminuria are less likely to have myeloma cast nephropathy but more likely to have myeloma-induced amyloidosis, light chain deposition disease, and heavy chain deposition disease.
- Patients with excess Bence-Jones proteinuria are at high risk of developing myeloma cast nephropathy.
- The risk for myeloma cast nephropathy increases significantly once the daily excretion of Bence-Jones protein exceeds 1 gram.[63]
Treatment / Management
Primary hyperoxaluria type 1 (PH1) therapy includes hyperhydration, using calcium oxalate crystallization, thiamine inhibitors, and pyridoxine. Pyridoxine effectively lowers oxalate levels in subgroups of PH1 patients with specific mutant alleles of the AGXT gene.[17](B3)
Treatment starts with pyridoxine at 5 mg/kg and then measuring urinary oxalate levels to assess the patient's response within two weeks of starting therapy. Patients should have at least a 30% reduction of urinary oxalate levels after three months of treatment to be considered pyridoxine responsive.[64] About 30% of patients with PH1 will respond to this treatment.[65] Pyridoxine should be stopped in patients who do not respond. The dosage needs to be titrated in patients who respond to the lowest possible effective dose but may be increased to a maximum of 20 mg/kg.[64][66] Patients on pyridoxine therapy should be monitored regularly for possible neurotoxicity.[3] Pyridoxine has no role in the treatment of PH2 and PH3.(B2)
Lumisaran was approved by the FDA in 2020 to treat primary hyperoxaluria type 1; it is an RNA interference (RNAi) agent that degrades the messenger RNA for hepatic glycolate oxidase. Glycolate oxidase is an enzyme that converts glycolate to glyoxylate, a precursor for oxalate production. Lumisaran decreases oxalate production by decreasing the amount of glyoxylate available to hepatic cells. It was shown to decrease urinary oxalate excretion by 65% after six months of treatment in the placebo-controlled phase 3 study ILLUMINATE-A; however, no change in eGFR was demonstrated in this short-term study. Similar results were noted in other phase-3 controlled studies, ILLUMINATE-B and ILLUMINATE-C. Early data from all the Lumisaran trials suggest that this drug can significantly decrease the risk of crystalluria and stabilize kidney function.[16][17][25](B3)
Stiripentol, an anti-epileptic agent, has been found to inhibit lactate dehydrogenase isoenzyme 5, which converts glyoxalate to oxalate. Nedosiran, which blocks the conversion of glyoxalate to oxalate through RNA interference, is being studied in PH1 and PH2 patients. Interestingly, it was shown to be effective only in PH1 patients, as no improvement in oxalate levels was noted in patients with PH2. This is thought to be secondary to the GRHPR enzyme being present in other tissues besides the liver. Liver transplantation may someday be avoided for primary hyperoxaluria patients if these and similar agents prove to be clinically successful.[16][17] Clinicians should remember that liver transplants have no role in decreasing the systemic oxalate load.(B3)
Renal replacement therapy should be considered in CKD stage IV and stage V patients even before the development of uremia to minimize systemic oxalosis. Aggressive hemodialysis should be done pretransplantation to decrease the systemic oxalate load in patients awaiting a renal transplant. Predialysis oxalate levels of 50 to 70 μmol/L should be targeted with aggressive dialysis: high flux and high-frequency hemodialysis should be the modality of renal replacement therapy, and the addition of nocturnal peritoneal dialysis can be considered in selected patients.[17] (B3)
Liver transplantation is the only known curative treatment for this condition. Patients who progress to CKD require combined liver and kidney transplantation. Ideally, this should be performed prior to the development of systemic oxalosis, as patients with a high systemic oxalate load carry the risk of crystal deposition in the transplanted kidney. In selected cases, continuous renal replacement therapy can be considered to decrease the systemic oxalate load during and immediately post-transplant.[16][17](B3)
Secondary oxalate nephropathy treatment hinges on sustaining sufficient hydration to generate more than 2 to 3 liters of daily urine output. Patients should be maintained on a low oxalate, high calcium, low-fat diet. Calcium citrate supplements are given with the most common high oxalate daily meals (usually lunch and dinner) to decrease intestinal oxalate absorption through increased gastrointestinal oxalate-calcium binding. Optimization of urinary crystallization inhibitors, like potassium citrate, is also helpful.[16]
Studies done to assess the efficacy of oral Oxalobacter formigenes and other probiotics in decreasing oxaluria did not show improved outcomes. A pilot study of an orally administered recombinant oxalate decarboxylase enzyme (which degrades oxalate), known as reloxilase, has shown promising results in patients with CKD and is being studied in a larger randomized control trial.[67] (B3)
Calcium phosphate deposition disease therapy includes optimized hydration, low sodium intake, potassium citrate supplementation, maintaining a normal calcium diet, and treating the disorder's underlying cause.[3][4]
2,8, DHA crystalluria treatment involves using a xanthine oxidase inhibitor, such as allopurinol or febuxostat, to overcome the APRT deficiency that causes this disorder.[68] Both agents effectively reduce 2,8 DHA excretion in patients with an APRT deficiency. A recent study showed that febuxostat is more effective in reducing the excretion of 2,8 DHA than allopurinol. However, both drugs are equally effective in decreasing the progression of CKD in this disorder.[69](A1)
Cystinosis therapy involves life-long oral cysteamine. The drug enhances cystine clearance, the dosing of which is guided by leukocyte cystine levels.[22] Cysteamine enters the lysosomes and breaks down the cystine into cysteine and cysteine-cysteamine disulfide, which are removed by the cysteine and cysteine-cysteamine transporters, respectively.[70] (B3)
Cysteamine treatment is not curative, and patients can have progressive renal failure even with long-term therapy. Long-term and early initiation of cysteamine treatment is associated with improvement in renal and extrarenal outcomes. While long-term cysteamine treatment can delay the progression to ESRD, it does not have any significant impact on renal Fanconi syndrome.[70] Gene therapy treatment is being investigated for the treatment and possible cure of this disease.[3][4](B3)
Cystinuria causes urinary cystine stones with characteristic hexagonal-shaped cystine crystals in the urine. Cystine stones are very dense and relatively resistant to standard nephrolithiasis treatment such as lithotripsy, so laser therapy is usually preferred. Cystine urinary crystals are increasingly soluble as the urinary pH increases, so treatment includes raising the urine pH to 7.5 or higher.[29] Very high hydration levels are needed to generate the recommended 3 liters of urine in 24 hours. Tiopronin is used as a cystine binder, which makes it far more soluble. For more information on cystine and cystinuria, see our companion Statpearls reference article on "Cystinuria."[29]
Acute urate nephropathy therapy primarily consists of aggressive hydration, urinary alkalization, and using xanthine oxidase inhibitors or recombinant urate oxidase, which converts the urate to water-soluble allantoin. While both treatment options can be used prophylactically, recombinant urate oxidase is particularly useful even after the development of acute urate nephropathy.[30][31](A1)
Multiple myeloma/paraproteinemia should be treated initially with supportive care, including hydration, avoidance of nephrotoxic medications, and treatment of any associated anemia or hypercalcemia. All patients with paraproteinemia-related crystalline nephropathy should get standard chemotherapy to reduce monoclonal paraprotein production.
Standard first-line treatment of newly diagnosed multiple myeloma is the combination of an immunomodulatory drug (lenalidomide), a proteasome inhibitor (bortezomib), and steroids (dexamethasone).[71](A1)
- Proteasome inhibitors have been shown to improve survival in multiple myeloma across multiple randomized trials and do not require dosage adjustment for renal impairment.
- Proteasome inhibitors rapidly reduce free light chain concentrations and inhibit the NF-kB inflammatory pathway, thereby decreasing tubular inflammation and apoptosis.
- Adding monoclonal anti-CD38 antibodies (daratumumab) to the standard of care has improved the complete remission rates in the GRIFFIN and CASSIOPEIA trials.[73][74] (A1)
- Daratumumab is also an acceptable substitute for patients who cannot tolerate bortezomib for any reason, such as peripheral neuropathy.[75] (A1)
Hemodialysis or peritoneal dialysis should be utilized in multiple myeloma patients who demonstrate or develop significant renal failure along with traditional indications such as uremia, hyperkalemia, worsening metabolic acidosis, volume overload, and pericarditis.
Autologous stem cell transplants are recommended and should be strongly considered in acceptable patients after 3 to 6 months of induction therapy.[9]
Extracorporeal light chain removal has long been entertained as an adjuvant treatment for myeloma cast nephropathy. Therapeutic plasma exchange involves the removal of harmful substances in the plasma. Plasma exchange is also known as single filter plasmapheresis, a modality in which blood is separated into its cellular components and plasma. The plasma with harmful proteins is discarded and replaced with albumin or fresh frozen plasma.
Two small randomized controlled trials on plasma exchange's role in multiple myeloma have shown conflicting results. The largest randomized controlled trial by Clark et al. had only 97 patients.[76] These patients were randomized into two groups of conventional chemotherapy and plasmapheresis in addition to the standard chemotherapy. The plasmapheresis group had up to 7 plasma exchanges on top of conventional chemotherapy. Plasma exchange was not shown to be beneficial in improving the primary composite outcome of reducing death and promoting renal recovery. This study has several important limitations as bortezomib chemotherapy was not used in these patients, and they did not have biopsy-confirmed myeloma cast nephropathy.[76](A1)
The use of plasma exchange remains controversial in the treatment of multiple myeloma. Currently, plasma exchange can be considered in patients with severe renal failure and very high levels of Bence-Jones proteinuria, as well as an adjunctive treatment in the management of crystaloglobulinemia.
High cut-off hemodialysis has also been suggested as another means of extracorporeal free light chain removal. A dialysis filter with a large-bore size of 10 nm is used with dialysis over weeks to remove free light chain immunoglobulins. HCO-HD (high-cutoff hemodialysis) membrane is capable of removing 90% of free light chains over three weeks and has beneficial effects in a single-center open-label study.[77] However, multiple randomized trials, such as the Eulite trial, were subsequently conducted to verify this benefit and could not confirm or reproduce these results; therefore, this treatement is not currently recommended.[78] (A1)
Differential Diagnosis
The differential diagnosis of primary hyperoxaluria includes PH2, PH3, hyperparathyroidism, and other causes of nephrocalcinosis as well as all the various causes of secondary hyperoxaluria.
The differential diagnosis of cystinosis, Dent disease, and Lowe's syndrome includes other causes of Fanconi syndrome, such as tyrosinemia, galactosemia, glycogen storage diseases, Wilsons disease, hereditary fructose intolerance, multiple mitochondrial disorders causing renal proximal tubular dysfunction, heavy metal toxicity from lead, cadmium, Sjögren syndrome, multiple myeloma and medications such as tenofovir, ifosfamide, cisplatin, sodium valproate, carbonic anhydrase inhibitors, topiramate, and aminoglycoside antibiotics.
The differential diagnosis of myeloma cast nephropathy includes amyloidosis, light chain deposition disease, heavy chain deposition disease, membranoproliferative glomerulonephritis, and hypercalcemia-induced AKI. Crystaloglobulinemia has a differential diagnosis including cryoglobulinemic vasculitis as well as other paraproteinemias.
Drug-induced crystalline nephropathy has a differential diagnosis that includes drug-induced interstitial nephritis, drug-induced tubular toxicity, and acute tubular necrosis.
Prognosis
Primary hyperoxaluria is associated with high morbidity and mortality in infants, but overall mortality has decreased over the past few years. Patients carry a high risk for progression to CKD and ESRD. Unfortunately, many patients tend to have delayed and missed diagnoses; some patients are diagnosed with this disease only after they develop allograft renal dysfunction.[56]
Patients with acute ingestion-associated calcium oxalate nephropathy generally have a good prognosis, while those with oxalate nephropathy due to enteric hyperoxaluria generally develop advanced and progressive renal disease.
2,8 DHA crystalluria's diagnosis is often delayed and missed. Patients develop recurrent nephrolithiasis, crystalluria, progressive renal failure, and ESRD without appropriate diagnosis and treatment. Many patients are diagnosed with this disease only after they develop allograft renal dysfunction. Sometimes patients are initially diagnosed with uric acid nephrolithiasis based on the radiolucency of 2,8 DHA crystals or are misdiagnosed as calcium oxalate crystallopathy based on the microscopic appearance. Patients with an early, accurate diagnosis and aggressive treatment with high-dose xanthine oxidase inhibitor therapy have a better prognosis and can avoid progression to ESRD.[68][69]
Long-term cysteamine treatment for cystinosis is associated with marked improvement in growth retardation and other endocrine disorders. Cystine accumulation in the brain can be associated with multiple structural brain abnormalities, cognitive impairment, and learning difficulties. Cysteamine treatment also improves multiple neurological symptoms and distal myopathy. This test is usually performed perinatally, and early initiation of treatment and treatment compliance are crucial to optimal outcomes and therapeutic success.[23][70]
Patients with myeloma-related proximal tubular crystalline nephropathy and crystalline histiocytosis have a slowly progressive renal disease and a reasonably good prognosis. These patients often tend to have low-grade multiple myeloma. The overall prognosis of multiple myeloma has improved significantly over the years.
Patients with myeloma cast nephropathy and crystaloglobulinemia tend to have a worse renal prognosis, ultimately requiring long-term renal replacement therapy.
Patients with drug-induced crystalluria generally have a good prognosis with renal recovery when there is early identification and termination of the culprit drug. However, a delay in the diagnosis and treatment of drug-induced crystalluria can cause irreversible renal dysfunction.
Complications
Patients with primary hyperoxaluria, secondary hyperoxaluria, and 2,8 DHA crystalluria are at increased risk for nephrolithiasis. These patients can develop acute kidney injury, obstructive uropathy causing urosepsis, progressive CKD, and ESRD.
Patients with PH1 have recurrent calcium oxalate kidney stones, rapidly progressive chronic kidney disease, and can progress to ESRD. The median onset of ESRD is 5.5 years, and most develop renal failure in the first three decades of life. Children with PH1 can develop complications such as growth retardation, failure to thrive, increased fractures, pancytopenia, hypothyroidism, and vascular calcifications leading to arterial insufficiency and heart block. Cardiac and vascular complications are commonly seen in adults.
Patients with cystinosis, Dent disease types 1 and 2, Lowe syndrome, and paraproteinemia-associated proximal tubular dysfunction can develop Fanconi syndrome. This has many concomitant complications, such as metabolic acidosis, hypophosphatemia, volume depletion, acute kidney injury, and nephrolithiasis. Children with Fanconi syndrome can develop rickets and growth retardation.
Patients with cystinosis can develop progressive renal failure, ESRD, hypothyroidism, and vacuolar myopathy, contributing to restrictive lung disease and increased risk for aspiration pneumonia, growth retardation, fractures, photophobia, hypogonadism, blindness, and exocrine and endocrine pancreatic insufficiency.[79]
Complications associated with myeloma cast nephropathy include progressive renal failure, multiple other extrarenal complications such as hypercalcemia, anemia, vertebral compression fractures, compressive myelopathy, nerve root compression, peripheral neuropathy, venous thromboembolism, and increased risk for infections.[80]
Deterrence and Patient Education
Patient education is critically important to optimize outcomes. The importance of compliance with treatment, monitoring, and follow-ups are critical to minimize renal damage. Genetic counseling is important in many of these disease entities with known inheritance patterns.
Enhancing Healthcare Team Outcomes
Crystalline nephropathies are increasingly recognized as a potential cause of progressive CKD and ESRD. Patient outcomes can be improved by recognizing primary hyperoxaluria as a cause of recurrent calcium oxalate stone disease, oxalate nephropathy, progressive CKD, and ESRD, even in adults. Some of these patients may be candidates for medical therapy with pyridoxine, lumisaran, stiripentol, or nedosiran.[16]
Nephrologists should collaborate with renal pathologists to diagnose patients with adenine phosphoribosyl transferase deficiency causing 2,8- DHA crystalluria and stones resulting in CKD and ESRD, which is treatable with xanthine oxidase inhibitors.[81]
Paraproteinemias cause various types of crystalline nephropathies. The outcomes can be improved with timely diagnosis and treatment, as signs of proximal tubular dysfunction may be the only identifiable clinical manifestation in a patient with paraproteinemia. Oncology input is crucial to the diagnosis of these conditions.
Patient outcomes can be improved with a thorough history of the patient's dietary habits, timely examination of urine sediment, and close collaboration with nephrologists, primary care physicians, urologists, pediatricians, genetic counselors, oncologists, dietitians, nurses, care coordinators, and renal pathologists in patients suspected of having crystallopathy.
Media
(Click Image to Enlarge)
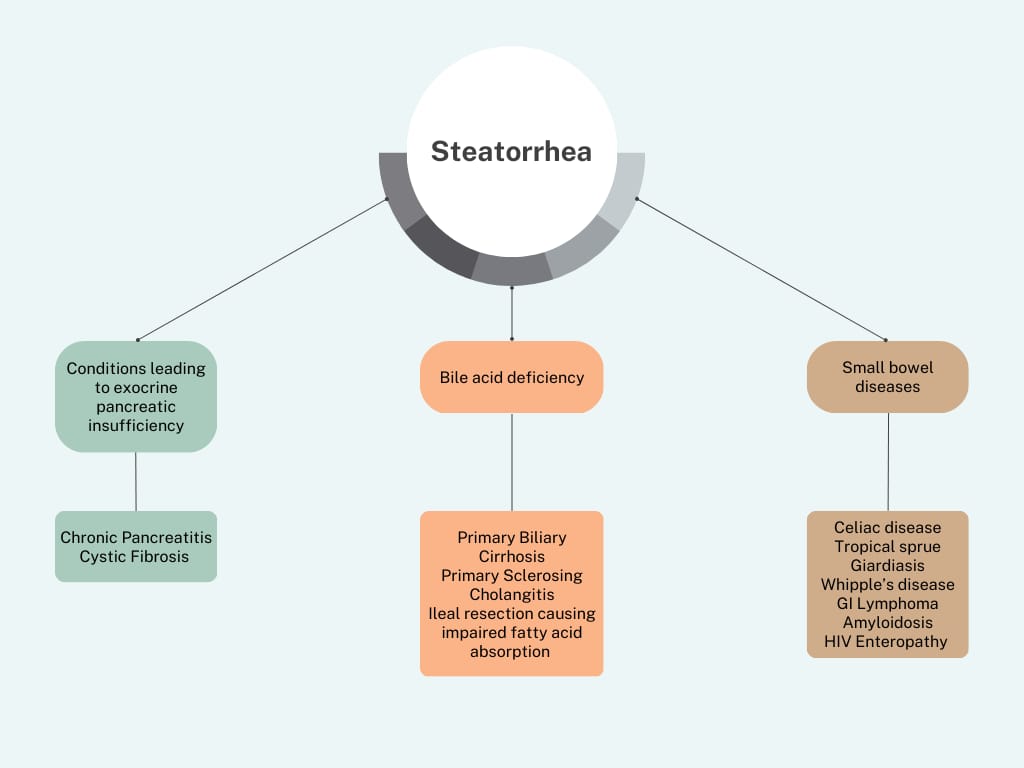
Causes of steatorrhea flow sheet
Contributed by Prathap Simhadri MD
(Click Image to Enlarge)
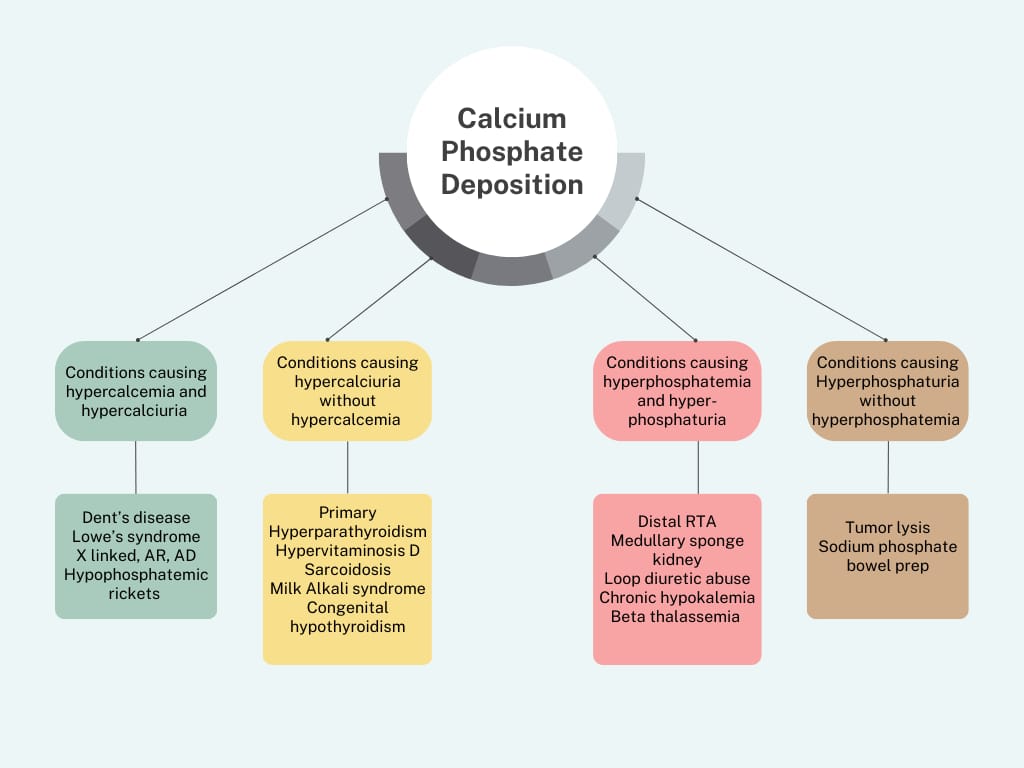
Causes of Calcium phosphate deposition flow sheet
Prathap Simhadri MD
(Click Image to Enlarge)
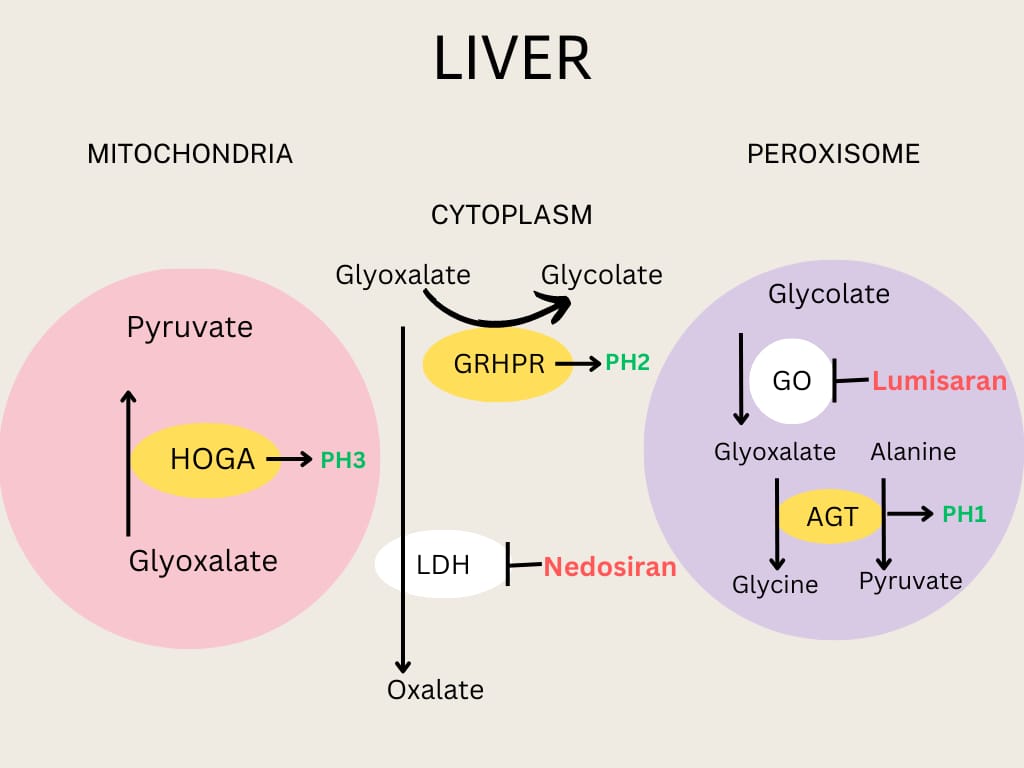
Causes of Primary Hyperoxaluria
contributed by Prathap Simhadri MD
(Click Image to Enlarge)
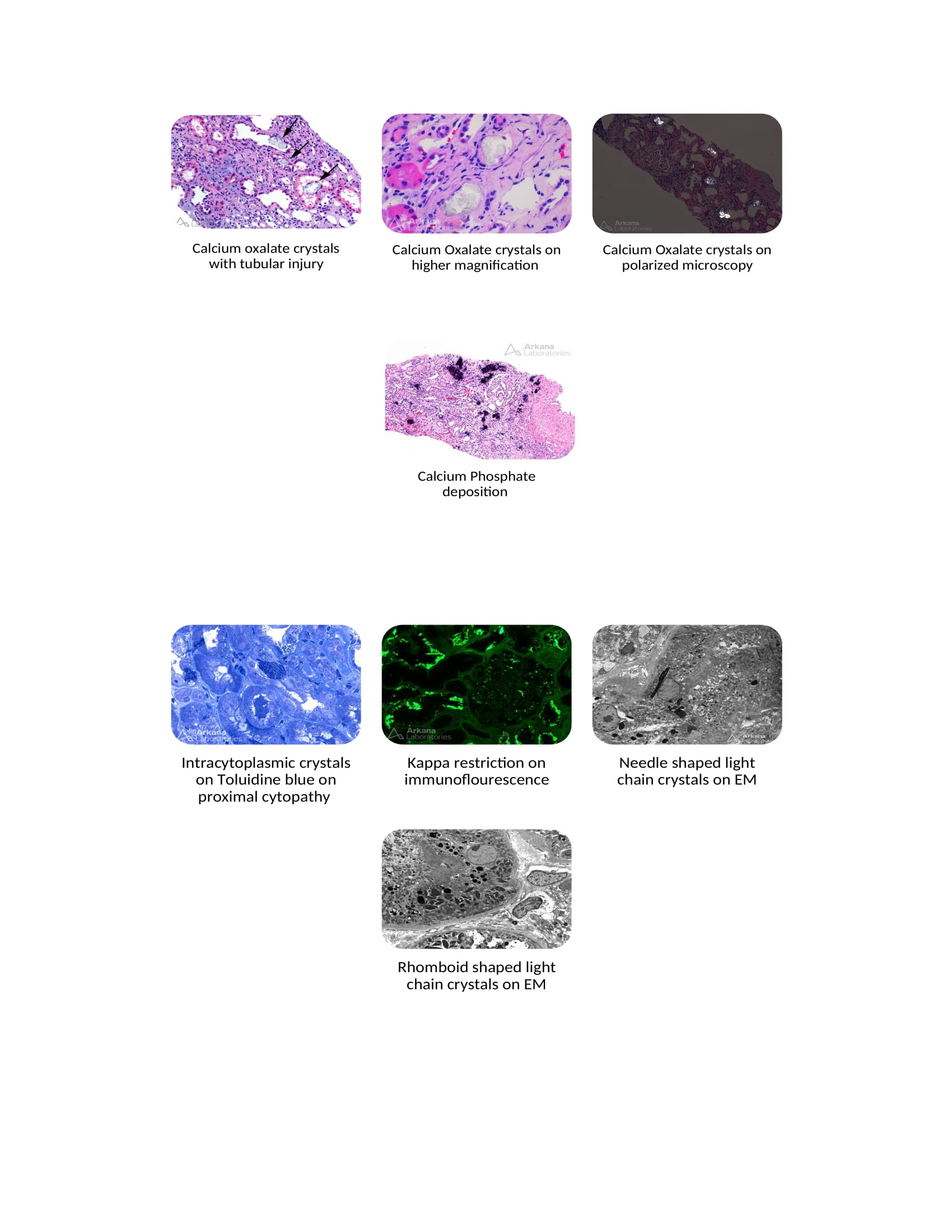
These pictures display the calcium oxalate crystals on renal biopsy specimens and light chain crystals on Proximal convoluted tubules
Used with permission from Arkanana Pathology Library
(Click Image to Enlarge)
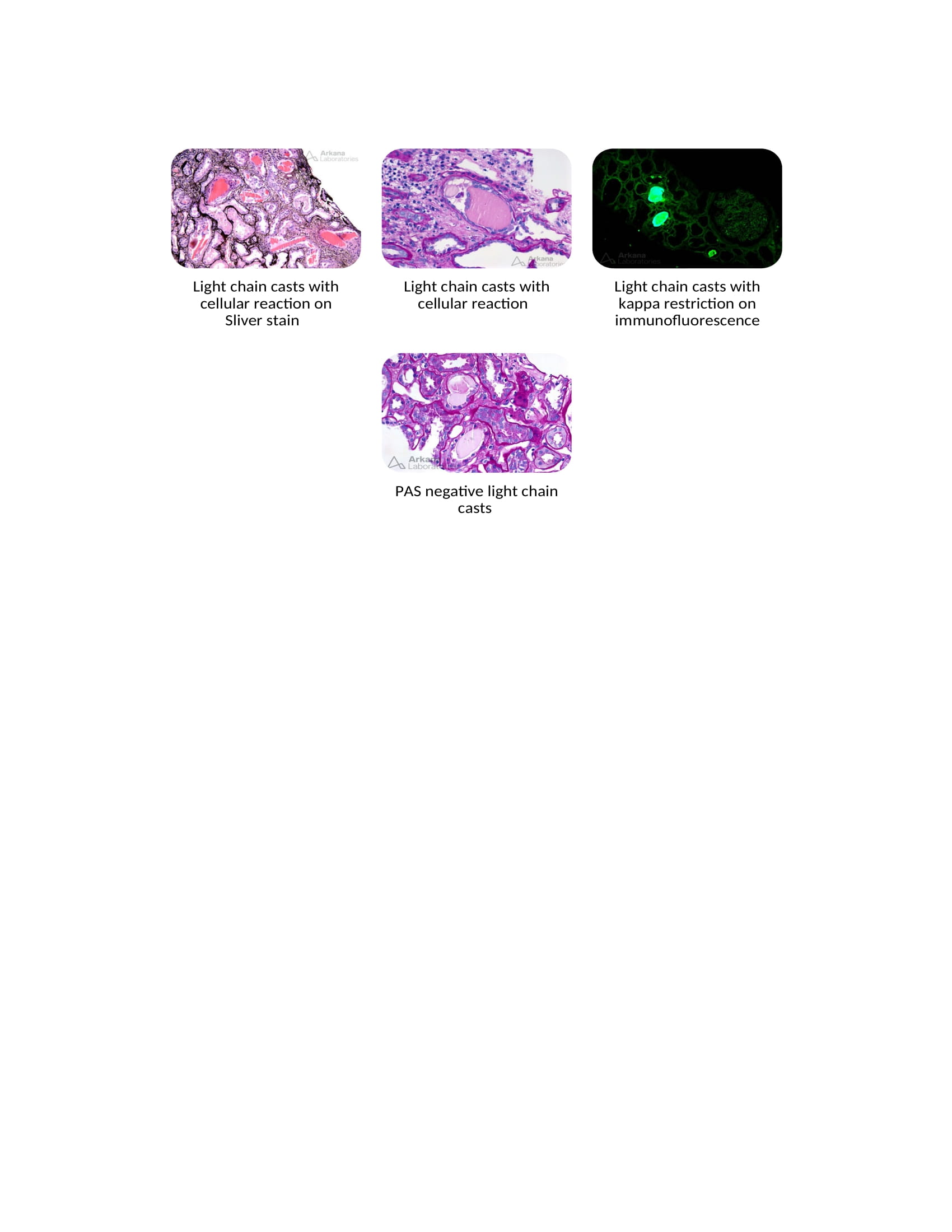
Light chain cast nephropathy slides
Used with Permission from Arkana Pathology Library
References
Rosenstock JL, Joab TMJ, DeVita MV, Yang Y, Sharma PD, Bijol V. Oxalate nephropathy: a review. Clinical kidney journal. 2022 Feb:15(2):194-204. doi: 10.1093/ckj/sfab145. Epub 2021 Aug 12 [PubMed PMID: 35145635]
Demoulin N, Aydin S, Gillion V, Morelle J, Jadoul M. Pathophysiology and Management of Hyperoxaluria and Oxalate Nephropathy: A Review. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2022 May:79(5):717-727. doi: 10.1053/j.ajkd.2021.07.018. Epub 2021 Sep 9 [PubMed PMID: 34508834]
Perazella MA, Herlitz LC. The Crystalline Nephropathies. Kidney international reports. 2021 Dec:6(12):2942-2957. doi: 10.1016/j.ekir.2021.09.003. Epub 2021 Sep 17 [PubMed PMID: 34901567]
Herlitz LC, D'Agati VD, Markowitz GS. Crystalline nephropathies. Archives of pathology & laboratory medicine. 2012 Jul:136(7):713-20. doi: 10.5858/arpa.2011-0565-RA. Epub [PubMed PMID: 22742545]
Elmonem MA, Veys KR, Soliman NA, van Dyck M, van den Heuvel LP, Levtchenko E. Cystinosis: a review. Orphanet journal of rare diseases. 2016 Apr 22:11():47. doi: 10.1186/s13023-016-0426-y. Epub 2016 Apr 22 [PubMed PMID: 27102039]
Kowalczyk NS, Zisman AL. Cystinuria: Review of a Life-long and Frustrating Disease. The Yale journal of biology and medicine. 2021 Dec:94(4):681-686 [PubMed PMID: 34970106]
Runolfsdottir HL, Palsson R, Agustsdottir IM, Indridason OS, Edvardsson VO. Kidney Disease in Adenine Phosphoribosyltransferase Deficiency. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2016 Mar:67(3):431-8. doi: 10.1053/j.ajkd.2015.10.023. Epub 2015 Dec 25 [PubMed PMID: 26724837]
Rajkumar SV. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. American journal of hematology. 2022 Aug:97(8):1086-1107. doi: 10.1002/ajh.26590. Epub 2022 May 23 [PubMed PMID: 35560063]
Cowan AJ, Green DJ, Kwok M, Lee S, Coffey DG, Holmberg LA, Tuazon S, Gopal AK, Libby EN. Diagnosis and Management of Multiple Myeloma: A Review. JAMA. 2022 Feb 1:327(5):464-477. doi: 10.1001/jama.2022.0003. Epub [PubMed PMID: 35103762]
Faye RB, Piraccini E. Vulvodynia. StatPearls. 2024 Jan:(): [PubMed PMID: 28613543]
Greenstein A, Militscher I, Chen J, Matzkin H, Lessing JB, Abramov L. Hyperoxaluria in women with vulvar vestibulitis syndrome. The Journal of reproductive medicine. 2006 Jun:51(6):500-2 [PubMed PMID: 16846091]
Solomons CC, Melmed MH, Heitler SM. Calcium citrate for vulvar vestibulitis. A case report. The Journal of reproductive medicine. 1991 Dec:36(12):879-82 [PubMed PMID: 1816400]
Level 3 (low-level) evidenceHarlow BL, Abenhaim HA, Vitonis AF, Harnack L. Influence of dietary oxalates on the risk of adult-onset vulvodynia. The Journal of reproductive medicine. 2008 Mar:53(3):171-8 [PubMed PMID: 18441720]
Level 2 (mid-level) evidenceAwad-Igbaria Y, Palzur E, Nasser M, Vieira-Baptista P, Bornstein J. Changes in the Vaginal Microbiota of Women With Secondary Localized Provoked Vulvodynia. Journal of lower genital tract disease. 2022 Oct 1:26(4):339-344. doi: 10.1097/LGT.0000000000000689. Epub 2022 Aug 6 [PubMed PMID: 35943448]
Yang Y, Sharma PD, Nair V, Jhaveri KD, Malieckal DA, Wanchoo R, Rosenstock JL, Bijol V. Kidney oxalate crystal deposition in adult patients: A relatively common finding . Clinical nephrology. 2020 May:93(5):243-250. doi: 10.5414/CN109980. Epub [PubMed PMID: 32101518]
Shah A, Leslie SW, Ramakrishnan S. Hyperoxaluria. StatPearls. 2024 Jan:(): [PubMed PMID: 32644413]
Groothoff JW, Metry E, Deesker L, Garrelfs S, Acquaviva C, Almardini R, Beck BB, Boyer O, Cerkauskiene R, Ferraro PM, Groen LA, Gupta A, Knebelmann B, Mandrile G, Moochhala SS, Prytula A, Putnik J, Rumsby G, Soliman NA, Somani B, Bacchetta J. Clinical practice recommendations for primary hyperoxaluria: an expert consensus statement from ERKNet and OxalEurope. Nature reviews. Nephrology. 2023 Mar:19(3):194-211. doi: 10.1038/s41581-022-00661-1. Epub 2023 Jan 5 [PubMed PMID: 36604599]
Level 3 (low-level) evidenceBuysschaert B, Aydin S, Morelle J, Gillion V, Jadoul M, Demoulin N. Etiologies, Clinical Features, and Outcome of Oxalate Nephropathy. Kidney international reports. 2020 Sep:5(9):1503-1509. doi: 10.1016/j.ekir.2020.06.021. Epub 2020 Jul 2 [PubMed PMID: 32954074]
Markowitz GS, Nasr SH, Klein P, Anderson H, Stack JI, Alterman L, Price B, Radhakrishnan J, D'Agati VD. Renal failure due to acute nephrocalcinosis following oral sodium phosphate bowel cleansing. Human pathology. 2004 Jun:35(6):675-84 [PubMed PMID: 15188133]
Level 3 (low-level) evidenceKunou M, Yamaguchi M, Takahashi H, Kimura Y, Watanabe N, Ito M, Sugiyama H, Iwagaitsu S, Nobata H, Kinashi H, Katsuno T, Banno S, Ito Y, Ishimoto T. A case of 2,8-DHA crystalline nephropathy caused by adenine phosphoribosyltransferase deficiency: diagnosis and treatment. CEN case reports. 2023 Aug:12(3):329-334. doi: 10.1007/s13730-022-00768-1. Epub 2022 Dec 28 [PubMed PMID: 36576711]
Level 3 (low-level) evidenceKalatzis V, Cherqui S, Antignac C, Gasnier B. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. The EMBO journal. 2001 Nov 1:20(21):5940-9 [PubMed PMID: 11689434]
Level 3 (low-level) evidenceHohenfellner K, Zerell K, Haffner D. Cystinosis. Klinische Monatsblatter fur Augenheilkunde. 2023 Mar:240(3):251-259. doi: 10.1055/a-2022-8522. Epub 2023 Mar 28 [PubMed PMID: 36977426]
Cherqui S, Courtoy PJ. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nature reviews. Nephrology. 2017 Feb:13(2):115-131. doi: 10.1038/nrneph.2016.182. Epub 2016 Dec 19 [PubMed PMID: 27990015]
Level 3 (low-level) evidenceHohenfellner K, Elenberg E, Ariceta G, Nesterova G, Soliman NA, Topaloglu R. Newborn Screening: Review of its Impact for Cystinosis. Cells. 2022 Mar 25:11(7):. doi: 10.3390/cells11071109. Epub 2022 Mar 25 [PubMed PMID: 35406673]
Scott LJ, Keam SJ. Lumasiran: First Approval. Drugs. 2021 Feb:81(2):277-282. doi: 10.1007/s40265-020-01463-0. Epub [PubMed PMID: 33405070]
Yarlagadda SG, Perazella MA. Drug-induced crystal nephropathy: an update. Expert opinion on drug safety. 2008 Mar:7(2):147-58. doi: 10.1517/14740338.7.2.147. Epub [PubMed PMID: 18324877]
Level 3 (low-level) evidenceChandra M, Stokes MB, Kaskel F. Multinucleated podocytes: a diagnostic clue to cystinosis. Kidney international. 2010 Nov:78(10):1052. doi: 10.1038/ki.2010.341. Epub [PubMed PMID: 21030980]
Level 3 (low-level) evidenceEmma F, Montini G, Pennesi M, Peruzzi L, Verrina E, Goffredo BM, Canalini F, Cassiman D, Rossi S, Levtchenko E. Biomarkers in Nephropathic Cystinosis: Current and Future Perspectives. Cells. 2022 Jun 4:11(11):. doi: 10.3390/cells11111839. Epub 2022 Jun 4 [PubMed PMID: 35681534]
Level 3 (low-level) evidenceLeslie SW, Sajjad H, Nazzal L. Cystinuria. StatPearls. 2024 Jan:(): [PubMed PMID: 29262245]
Mei Y, Dong B, Geng Z, Xu L. Excess Uric Acid Induces Gouty Nephropathy Through Crystal Formation: A Review of Recent Insights. Frontiers in endocrinology. 2022:13():911968. doi: 10.3389/fendo.2022.911968. Epub 2022 Jul 14 [PubMed PMID: 35909538]
Gonçalves DLN, Moreira TR, da Silva LS. A systematic review and meta-analysis of the association between uric acid levels and chronic kidney disease. Scientific reports. 2022 Apr 15:12(1):6251. doi: 10.1038/s41598-022-10118-x. Epub 2022 Apr 15 [PubMed PMID: 35428828]
Level 1 (high-level) evidenceKC M, Leslie SW. Uric Acid Nephrolithiasis. StatPearls. 2024 Jan:(): [PubMed PMID: 32809561]
Kaur P, Bhatt H. Hyperuricosuria. StatPearls. 2024 Jan:(): [PubMed PMID: 32965872]
George C, Leslie SW, Minter DA. Hyperuricemia. StatPearls. 2024 Jan:(): [PubMed PMID: 29083565]
Sanders PW. Mechanisms of light chain injury along the tubular nephron. Journal of the American Society of Nephrology : JASN. 2012 Nov:23(11):1777-81. doi: 10.1681/ASN.2012040388. Epub 2012 Sep 20 [PubMed PMID: 22997259]
Markowitz GS. Dysproteinemia and the kidney. Advances in anatomic pathology. 2004 Jan:11(1):49-63 [PubMed PMID: 14676640]
Level 3 (low-level) evidenceWu CK, Yang AH, Lai HC, Lin BS. Combined proximal tubulopathy, crystal-storing histiocytosis, and cast nephropathy in a patient with light chain multiple myeloma. BMC nephrology. 2017 May 25:18(1):170. doi: 10.1186/s12882-017-0584-8. Epub 2017 May 25 [PubMed PMID: 28545410]
Sapkota S, Shaikh H. Non-Hodgkin Lymphoma. StatPearls. 2024 Jan:(): [PubMed PMID: 32644754]
Masood A, Ehsan H, Iqbal Q, Salman A, Hashmi H. Treatment of Light Chain Deposition Disease: A Systematic Review. Journal of hematology. 2022 Aug:11(4):123-130. doi: 10.14740/jh1038. Epub 2022 Aug 30 [PubMed PMID: 36118549]
Level 1 (high-level) evidenceSirac C, Batuman V, Sanders PW. The Proximal Tubule Toxicity of Immunoglobulin Light Chains. Kidney international reports. 2021 May:6(5):1225-1231. doi: 10.1016/j.ekir.2021.02.026. Epub 2021 Mar 3 [PubMed PMID: 34013100]
Sanders PW, Booker BB, Bishop JB, Cheung HC. Mechanisms of intranephronal proteinaceous cast formation by low molecular weight proteins. The Journal of clinical investigation. 1990 Feb:85(2):570-6 [PubMed PMID: 2298921]
Level 3 (low-level) evidenceZeisberg M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. Journal of the American Society of Nephrology : JASN. 2010 Nov:21(11):1819-34. doi: 10.1681/ASN.2010080793. Epub 2010 Sep 23 [PubMed PMID: 20864689]
Finkel KW, Cohen EP, Shirali A, Abudayyeh A, American Society of Nephrology Onco-Nephrology Forum. Paraprotein-Related Kidney Disease: Evaluation and Treatment of Myeloma Cast Nephropathy. Clinical journal of the American Society of Nephrology : CJASN. 2016 Dec 7:11(12):2273-2279. doi: 10.2215/CJN.01640216. Epub 2016 Aug 15 [PubMed PMID: 27526708]
Clark WF, Garg AX. Plasma exchange for myeloma kidney: cast(s) away? Kidney international. 2008 Jun:73(11):1211-3. doi: 10.1038/ki.2008.117. Epub [PubMed PMID: 18480853]
Leung N, Gertz MA, Zeldenrust SR, Rajkumar SV, Dispenzieri A, Fervenza FC, Kumar S, Lacy MQ, Lust JA, Greipp PR, Witzig TE, Hayman SR, Russell SJ, Kyle RA, Winters JL. Improvement of cast nephropathy with plasma exchange depends on the diagnosis and on reduction of serum free light chains. Kidney international. 2008 Jun:73(11):1282-8. doi: 10.1038/ki.2008.108. Epub 2008 Apr 2 [PubMed PMID: 18385667]
Level 2 (mid-level) evidenceGupta V, El Ters M, Kashani K, Leung N, Nasr SH. Crystalglobulin-induced nephropathy. Journal of the American Society of Nephrology : JASN. 2015 Mar:26(3):525-9. doi: 10.1681/ASN.2014050509. Epub 2014 Sep 4 [PubMed PMID: 25190731]
Level 3 (low-level) evidenceMobarki M, Papoudou-Bai A, Dumollard JM, Alhazmi AH, Musawi S, Madkhali MA, Muqri KY, Péoc'h M, Karpathiou G. Crystal-Storing Histiocytosis: The Iceberg of More Serious Conditions. Diagnostics (Basel, Switzerland). 2023 Jan 11:13(2):. doi: 10.3390/diagnostics13020271. Epub 2023 Jan 11 [PubMed PMID: 36673081]
Ungari M, Ghiringhelli P, Marchi G, Fisogni S, Lavazza A, Molteni A, Malberti F, Bertoni R, Trombatore M, Ferrero G, Gusolfino MD, Varotti E, Tanzi G, Manotti L. Combined renal proximal tubulopathy and crystal storing histiocytosis in a patient with κ light chain multiple myeloma. Pathologica. 2021 Aug:113(4):285-293. doi: 10.32074/1591-951X-154. Epub [PubMed PMID: 34463673]
Zhu L, Wang L, Shi H, Jiang L, Li X, Shao C, Yan Y, Dong B, Zou W, Zuo L. Combined crystal-storing histiocytosis, light chain proximal tubulopathy, and light chain crystalline podocytopathy in a patient with multiple myeloma: a case report and literature review. Renal failure. 2023 Dec:45(1):2145970. doi: 10.1080/0886022X.2022.2145970. Epub [PubMed PMID: 36632756]
Level 3 (low-level) evidenceYu XJ, Zhou XJ, Wang SX, Zhou FD, Zhao MH. Monoclonal light chain crystalline podocytopathy and tubulopathy associated with monoclonal gammopathy of renal significance: a case report and literature review. BMC nephrology. 2018 Nov 12:19(1):322. doi: 10.1186/s12882-018-1108-x. Epub 2018 Nov 12 [PubMed PMID: 30419839]
Level 3 (low-level) evidenceEymieux S, Miquelestorena-Standley E, Rabot N, Maisons V, Touchard G, Blanchard E. Crystalline podocytopathy and tubulopathy linked to kappa light chain deposits in a context of smoldering multiple myeloma. Clinical kidney journal. 2022 Feb:15(2):351-353. doi: 10.1093/ckj/sfab197. Epub 2021 Oct 6 [PubMed PMID: 35145650]
Leslie SW, Sajjad H, Murphy PB. Renal Calculi, Nephrolithiasis. StatPearls. 2024 Jan:(): [PubMed PMID: 28723043]
Patti L, Leslie SW. Acute Renal Colic. StatPearls. 2024 Jan:(): [PubMed PMID: 28613743]
Gilman TM, Boylen CT, Sattler FR. Pyrimethamine-sulfadiazine for treating Pneumocystis carinii pneumonia and toxoplasmosis in AIDS. Clinical pharmacy. 1989 Feb:8(2):84, 87 [PubMed PMID: 2645082]
Level 3 (low-level) evidenceGeorgiev VS. Management of toxoplasmosis. Drugs. 1994 Aug:48(2):179-88 [PubMed PMID: 7527323]
Ben-Shalom E, Garrelfs SF, Groothoff JW. Primary hyperoxaluria: the pediatric nephrologist's point of view. Clinical kidney journal. 2022 May:15(Suppl 1):i23-i28. doi: 10.1093/ckj/sfab231. Epub 2022 May 17 [PubMed PMID: 35592624]
Murad S, Eisenberg Y. ENDOCRINE MANIFESTATIONS OF PRIMARY HYPEROXALURIA. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2017 Dec:23(12):1414-1424. doi: 10.4158/EP-2017-0029. Epub 2017 Nov 16 [PubMed PMID: 29144803]
Bhasin B, Ürekli HM, Atta MG. Primary and secondary hyperoxaluria: Understanding the enigma. World journal of nephrology. 2015 May 6:4(2):235-44. doi: 10.5527/wjn.v4.i2.235. Epub [PubMed PMID: 25949937]
Level 3 (low-level) evidenceSoliman NA, Nabhan MM, Abdelrahman SM, Abdelaziz H, Helmy R, Ghanim K, Bazaraa HM, Badr AM, Tolba OA, Kotb MA, Eweeda KM, Fayez A. Clinical spectrum of primary hyperoxaluria type 1: Experience of a tertiary center. Nephrologie & therapeutique. 2017 May:13(3):176-182. doi: 10.1016/j.nephro.2016.08.002. Epub 2017 Feb 1 [PubMed PMID: 28161266]
Greed L, Willis F, Johnstone L, Teo S, Belostotsky R, Frishberg Y, Pitt J. Metabolite diagnosis of primary hyperoxaluria type 3. Pediatric nephrology (Berlin, Germany). 2018 Aug:33(8):1443-1446. doi: 10.1007/s00467-018-3967-6. Epub 2018 Apr 28 [PubMed PMID: 29705963]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Milliner DS, Harris PC, Sas DJ, Cogal AG, Lieske JC. Primary Hyperoxaluria Type 1. GeneReviews(®). 1993:(): [PubMed PMID: 20301460]
Keefe P, Bokhari SRA. Fanconi Syndrome. StatPearls. 2024 Jan:(): [PubMed PMID: 30521293]
Sathick IJ, Drosou ME, Leung N. Myeloma light chain cast nephropathy, a review. Journal of nephrology. 2019 Apr:32(2):189-198. doi: 10.1007/s40620-018-0492-4. Epub 2018 May 5 [PubMed PMID: 29730782]
Hoppe B, Martin-Higueras C. Inherited conditions resulting in nephrolithiasis. Current opinion in pediatrics. 2020 Apr:32(2):273-283. doi: 10.1097/MOP.0000000000000848. Epub [PubMed PMID: 31789978]
Level 3 (low-level) evidencevan Woerden CS, Groothoff JW, Wijburg FA, Annink C, Wanders RJ, Waterham HR. Clinical implications of mutation analysis in primary hyperoxaluria type 1. Kidney international. 2004 Aug:66(2):746-52 [PubMed PMID: 15253729]
Level 2 (mid-level) evidenceGupta A, Somers MJG, Baum MA. Treatment of primary hyperoxaluria type 1. Clinical kidney journal. 2022 May:15(Suppl 1):i9-i13. doi: 10.1093/ckj/sfab232. Epub 2022 May 17 [PubMed PMID: 35592620]
Pfau A, Grujic D, Keddis MT, Kausz AT, Lieske JC, Knauf F. Pilot study of reloxaliase in patients with severe enteric hyperoxaluria and hyperoxalemia. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2021 Apr 26:36(5):945-948. doi: 10.1093/ndt/gfaa379. Epub [PubMed PMID: 33416876]
Level 3 (low-level) evidenceRashid I, Verma A, Tiwari P, D'Cruz S. Adenine phosphoribosyl transferase deficiency leads to renal allograft dysfunction in kidney transplant recipients: a systematic review. Jornal brasileiro de nefrologia. 2022 Jul-Sep:44(3):403-416. doi: 10.1590/2175-8239-JBN-2021-0283en. Epub [PubMed PMID: 35635787]
Level 1 (high-level) evidenceEdvardsson VO, Runolfsdottir HL, Thorsteinsdottir UA, Sch Agustsdottir IM, Oddsdottir GS, Eiriksson F, Goldfarb DS, Thorsteinsdottir M, Palsson R. Comparison of the effect of allopurinol and febuxostat on urinary 2,8-dihydroxyadenine excretion in patients with Adenine phosphoribosyltransferase deficiency (APRTd): A clinical trial. European journal of internal medicine. 2018 Feb:48():75-79. doi: 10.1016/j.ejim.2017.10.007. Epub 2017 Dec 12 [PubMed PMID: 29241594]
Jézégou A, Llinares E, Anne C, Kieffer-Jaquinod S, O'Regan S, Aupetit J, Chabli A, Sagné C, Debacker C, Chadefaux-Vekemans B, Journet A, André B, Gasnier B. Heptahelical protein PQLC2 is a lysosomal cationic amino acid exporter underlying the action of cysteamine in cystinosis therapy. Proceedings of the National Academy of Sciences of the United States of America. 2012 Dec 11:109(50):E3434-43. doi: 10.1073/pnas.1211198109. Epub 2012 Nov 20 [PubMed PMID: 23169667]
Level 3 (low-level) evidenceDurie BGM, Hoering A, Abidi MH, Rajkumar SV, Epstein J, Kahanic SP, Thakuri M, Reu F, Reynolds CM, Sexton R, Orlowski RZ, Barlogie B, Dispenzieri A. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): a randomised, open-label, phase 3 trial. Lancet (London, England). 2017 Feb 4:389(10068):519-527. doi: 10.1016/S0140-6736(16)31594-X. Epub 2016 Dec 23 [PubMed PMID: 28017406]
Level 1 (high-level) evidenceKumar S, Flinn I, Richardson PG, Hari P, Callander N, Noga SJ, Stewart AK, Turturro F, Rifkin R, Wolf J, Estevam J, Mulligan G, Shi H, Webb IJ, Rajkumar SV. Randomized, multicenter, phase 2 study (EVOLUTION) of combinations of bortezomib, dexamethasone, cyclophosphamide, and lenalidomide in previously untreated multiple myeloma. Blood. 2012 May 10:119(19):4375-82. doi: 10.1182/blood-2011-11-395749. Epub 2012 Mar 15 [PubMed PMID: 22422823]
Level 1 (high-level) evidenceVoorhees PM, Kaufman JL, Laubach J, Sborov DW, Reeves B, Rodriguez C, Chari A, Silbermann R, Costa LJ, Anderson LD Jr, Nathwani N, Shah N, Efebera YA, Holstein SA, Costello C, Jakubowiak A, Wildes TM, Orlowski RZ, Shain KH, Cowan AJ, Murphy S, Lutska Y, Pei H, Ukropec J, Vermeulen J, de Boer C, Hoehn D, Lin TS, Richardson PG. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: the GRIFFIN trial. Blood. 2020 Aug 20:136(8):936-945. doi: 10.1182/blood.2020005288. Epub [PubMed PMID: 32325490]
Moreau P, Hulin C, Perrot A, Arnulf B, Belhadj K, Benboubker L, Béné MC, Zweegman S, Caillon H, Caillot D, Corre J, Delforge M, Dejoie T, Doyen C, Facon T, Sonntag C, Fontan J, Mohty M, Jie KS, Karlin L, Kuhnowski F, Lambert J, Leleu X, Macro M, Orsini-Piocelle F, Roussel M, Stoppa AM, van de Donk NWCJ, Wuillème S, Broijl A, Touzeau C, Tiab M, Marolleau JP, Meuleman N, Vekemans MC, Westerman M, Klein SK, Levin MD, Offner F, Escoffre-Barbe M, Eveillard JR, Garidi R, Ahmadi T, Krevvata M, Zhang K, de Boer C, Vara S, Kampfenkel T, Vanquickelberghe V, Vermeulen J, Avet-Loiseau H, Sonneveld P. Maintenance with daratumumab or observation following treatment with bortezomib, thalidomide, and dexamethasone with or without daratumumab and autologous stem-cell transplant in patients with newly diagnosed multiple myeloma (CASSIOPEIA): an open-label, randomised, phase 3 trial. The Lancet. Oncology. 2021 Oct:22(10):1378-1390. doi: 10.1016/S1470-2045(21)00428-9. Epub 2021 Sep 13 [PubMed PMID: 34529931]
Level 1 (high-level) evidenceFacon T, Kumar SK, Plesner T, Orlowski RZ, Moreau P, Bahlis N, Basu S, Nahi H, Hulin C, Quach H, Goldschmidt H, O'Dwyer M, Perrot A, Venner CP, Weisel K, Mace JR, Raje N, Tiab M, Macro M, Frenzel L, Leleu X, Ahmadi T, Wang J, Van Rampelbergh R, Uhlar CM, Tromp B, Delioukina M, Vermeulen J, Usmani SZ. Daratumumab, lenalidomide, and dexamethasone versus lenalidomide and dexamethasone alone in newly diagnosed multiple myeloma (MAIA): overall survival results from a randomised, open-label, phase 3 trial. The Lancet. Oncology. 2021 Nov:22(11):1582-1596. doi: 10.1016/S1470-2045(21)00466-6. Epub 2021 Oct 13 [PubMed PMID: 34655533]
Level 1 (high-level) evidenceClark WF, Stewart AK, Rock GA, Sternbach M, Sutton DM, Barrett BJ, Heidenheim AP, Garg AX, Churchill DN, Canadian Apheresis Group. Plasma exchange when myeloma presents as acute renal failure: a randomized, controlled trial. Annals of internal medicine. 2005 Dec 6:143(11):777-84 [PubMed PMID: 16330788]
Level 1 (high-level) evidenceHutchison CA, Bradwell AR, Cook M, Basnayake K, Basu S, Harding S, Hattersley J, Evans ND, Chappel MJ, Sampson P, Foggensteiner L, Adu D, Cockwell P. Treatment of acute renal failure secondary to multiple myeloma with chemotherapy and extended high cut-off hemodialysis. Clinical journal of the American Society of Nephrology : CJASN. 2009 Apr:4(4):745-54. doi: 10.2215/CJN.04590908. Epub 2009 Apr 1 [PubMed PMID: 19339414]
Level 3 (low-level) evidenceHutchison CA, Cockwell P, Moroz V, Bradwell AR, Fifer L, Gillmore JD, Jesky MD, Storr M, Wessels J, Winearls CG, Weisel K, Heyne N, Cook M. High cutoff versus high-flux haemodialysis for myeloma cast nephropathy in patients receiving bortezomib-based chemotherapy (EuLITE): a phase 2 randomised controlled trial. The Lancet. Haematology. 2019 Apr:6(4):e217-e228. doi: 10.1016/S2352-3026(19)30014-6. Epub 2019 Mar 11 [PubMed PMID: 30872075]
Level 1 (high-level) evidenceNesterova G, Gahl WA. Cystinosis: the evolution of a treatable disease. Pediatric nephrology (Berlin, Germany). 2013 Jan:28(1):51-9. doi: 10.1007/s00467-012-2242-5. Epub 2012 Aug 18 [PubMed PMID: 22903658]
Sullivan E, Smith LC, Falco AM. Multiple Myeloma: Cast Nephropathy, VTE, and Neurologic Complications. Journal of the advanced practitioner in oncology. 2013 Jan:4(1):37-46 [PubMed PMID: 25031979]
Jaffer A, Joyce A, Koenig P, Biyani CS. Adenine Phosphoribosyltransferase Deficiency: A Rare Cause of Recurrent Urolithiasis. Journal of endourology case reports. 2017:3(1):49-51. doi: 10.1089/cren.2017.0015. Epub 2017 Apr 1 [PubMed PMID: 28466077]
Level 3 (low-level) evidence