Continuing Education Activity
Absence seizures are classified as a type of generalized onset nonmotor seizure with clinical symptoms, including unresponsiveness and staring spells. Absence seizures are generally seen in children aged 5 to 15 years and occur in multiple genetic generalized epilepsies, including childhood absence epilepsy (CAE), juvenile absence epilepsy (JAE), and juvenile myoclonic epilepsy (JME). Atypical absence seizures have been reported in up to 60% of patients with Lennox-Gastaut syndrome. A classic EEG finding of a 3-Hz spike and wave discharges is found. Absence epilepsy is an electroclinical diagnosis (clinical presentation and EEG findings). While primarily affecting children, absence seizures can persist into adulthood, significantly impacting daily functioning and quality of life if left untreated.
This course aims to provide an in-depth understanding of absence seizures, a condition that often goes unrecognized or misdiagnosed. The clinical presentation, pathophysiology, diagnostic criteria, and treatment options for absence seizures are reviewed. This activity also addresses the unique electroencephalogram (EEG) findings characteristic of absence seizures and the differential diagnosis to distinguish them from other seizure types and nonepileptic events. Participants will gain insights into the latest evidence-based management strategies, including pharmacological interventions and nonpharmacological approaches, tailored to effectively control absence seizures. The role of an interprofessional team in managing this challenging neurological condition is highlighted.
Objectives:
Evaluate a patient presenting with staring spells for characteristics consistent with absence seizures.
Identify the first-line medications for absence epilepsy and associated adverse effects.
Assess approaches to counsel absence seizure patients and family members on safety precautions.
Collaborate with interprofessional team members to promptly diagnose, evaluate, and treat patients with absence seizures.
Introduction
Absence seizures are brief seizures characterized by a behavioral arrest correlating with generalized 3-Hz spike and wave discharges on electroencephalogram (EEG).[1] Absence seizures occur in multiple genetic generalized epilepsies, including childhood absence epilepsy (CAE), juvenile absence epilepsy (JAE), and juvenile myoclonic epilepsy (JME).[2] Genetic generalized epilepsies comprise about one-fourth of all epilepsies. Atypical absence seizures have been reported in up to 60% of patients with Lennox-Gastaut syndrome.
Historically, absence epilepsy was known as "pyknolepsy." This originates from the Greek term pyknos, meaning "very frequent" or "grouped."[3][4] The term "petit mal" seizure was previously used to describe an absence seizure, but the term is no longer encouraged. The International League Against Epilepsy Classification of 2017 defines absence seizures as "generalized nonmotor seizures."[5] However, this term is not entirely precise because, as discussed below, motor manifestations of absence epilepsy are frequently seen.[2]
Absence seizures have often been considered "benign" due to their nonconvulsive nature; however, up to 60% of children with absence seizures have severe neuropsychic manifestations, including disturbances in attention, cognition, memory, and mood.[6] Absence epilepsy affects a child's psychosocial and academic function and requires treatment.
Etiology
A genetic component exists for all generalized epilepsies and, specifically, for absence epilepsy. The inheritance pattern is not strictly autosomal recessive or dominant and is considered to be multifactorial and polygenic. In 1951, Lennox reported that 66% of monozygotic twins showed concordance for the EEG pattern of 3-Hertz spike and wave discharges. These can be voltage-gated (CACNA1H, CACNG3, CLCN2) or ligand-gated (CHRNA4, GABRA1, GABRB3, GABRG2, GRM4).[7] CAE is linked to many genetic variations, including at the following locations: [7]
- Calcium-channel genes: CACNA1H and CACNG3 (especially in Han Chinese) [8]
- GABA-A and GABA-B receptors: GABA is an inhibitory neurotransmitter highly involved in epileptogenesis. GABRA1, GABRB3, and GABRG2 are variants relating to CAE.[3][4][9]
- Glutamate receptor: Mutations in GRM4 alter glutamate and GABA regulation.
- µ-Opioid Receptor: OPRM1 is an opioid receptor where mutations can cause thalamic hyperexcitability.
- Solute transfer receptor: SLC6A3/DAT1 are 2 genes in this category encoding a dopamine transporter.
Also, some copy number variants (CNVs), for example, 15q11.2, 15q13.3, and 16p13.11 microdeletion, have been described in patients with CAE.[10] However, the mode of inheritance and the majority of genes implicated in CAE are still unknown.[4]
Epidemiology
The incidence of CAE is approximately 6.3 to 8.0 children per 100,000 individuals per year.[10][11][12] CAE is a common pediatric epilepsy syndrome. Among all cases of epilepsy in school-aged children, 10% to 17% are due to CAE.[1] The age of onset for CAE is typically between 4 and 10 years, with a peak between ages 5 and 7. Girls have CAE more frequently than boys; however, some of this evidence is conflicting.[4]
Pathophysiology
The pathophysiological mechanism of the development of absence seizures is not yet fully understood. However, the cortico-thalamic-cortical circuit is believed to play a significant role in the pathophysiology of absence seizures.[13]
Some of the neurons involved in the cortico-thalamic-cortical system include the following:
- Cortical glutamatergic neurons originating on cortical layer VI and projecting to the nucleus reticularis of the thalamus
- Thalamic relay neurons with excitatory projections to cortical pyramidal neurons
- Neurons from the thalamic nucleus reticularis containing inhibitory GABA-ergic projections that connect with other neurons from the same nucleus and with thalamic relay neurons. These neurons do not connect directly with the cortex.[4]
Neurons from the thalamic nucleus reticularis can fire in an oscillatory pattern (eg, rhythmic bursts involved in generating sleep spindles) or continuously in single spikes (tonic firing during wakefulness). Shifts between these 2 firing patterns in the thalamic nucleus reticularis are modulated by spikes in thalamocortical networks and neurons from the thalamic nucleus reticularis. These are mediated through low-threshold transient calcium channels known as T-type channels. After depolarization, T-type channels briefly allow calcium inflow before becoming inactivated. Reactivation requires a relatively long hyperpolarization facilitated by GABA-B receptors. Therefore, abnormal oscillatory rhythms can originate from T-type channel abnormality from increased GABA-B activity.[4]
As explained by the genetics of absence epilepsy, genes coding for T-type calcium channels and GABA receptors have been associated with the etiopathogenesis of this type of epilepsy. Medications that suppress T-type calcium channels, such as ethosuximide and valproate, are effective anti-absence drugs. Conversely, medications that increase GABA-B activity (eg, vigabatrin) exacerbate the frequency of absence seizures. In contrast, GABA-A agonists (eg, benzodiazepines) that preferentially enhance GABA-ergic activity in neurons from the thalamic nucleus reticularis can suppress absence seizures.[4]
More recent research on penicillin-induced models of epilepsy in cats favors the cerebellum's role in long-term electrical stimulation in absence epilepsy.[14]
History and Physical
The age of onset of CAE is usually between 4 and 10 years, with a peak between the ages of 5 and 7.[4] The onset of absence epilepsy before age 4 should raise concern for an underlying glucose transporter type 1 (GLUT1) deficiency.[4] About 10% to 15% of patients with CAE have a history of febrile seizures.[14]
Regarding the clinical presentation, family members and teachers usually describe brief spells in which the child has a loss of awareness, is unresponsive, and has a behavioral arrest. Bystanders often describe the patient during these spells as having “a blank stare.”[2] Episodes frequently occur, often 10 to 30 times throughout the day. Most children stop their activity completely, but some can continue it more slowly or unusually.[1] Some children have 3-Hz regular eyelid fluttering.[1][2] Clinical presentation can also vary in the same individual.[7]
Oral automatisms can also occur, especially with prolonged seizures or during hyperventilation. Children frequently have mild clonic or tonic movements in the first few seconds of the spell. Pallor is commonly reported.[1] Urinary incontinence is rare. Spells usually last between 4 and 30 seconds. Hyperventilation, a state of arousal, sleep deprivation, and medication use can affect the duration of the seizure.[1] These seizures are not preceded by an aura and do not have a postictal state.[13]
On physical examination, hyperventilation can trigger absence seizures. To test this, the examiner asks the child to blow repetitively for at least 2 minutes. Using a pinwheel or paper can be helpful because this encourages the child to cooperate more during testing. If hyperventilation is done successfully, the patient may develop seizures that can be seen clinically or on EEG. There is some evidence that absence seizures are more easily provoked when the person is in a sitting position.[15]
For JAE, the age of onset is classically between 10 and 19 years, with a peak at 15 years.[4] Seizures are less frequent than in CAE but tend to last longer.[3][4]
Absence status consists of generalized, nonconvulsive seizures characterized by impairment of awareness and intermittently has other manifestations such as automatisms or subtle myoclonic, tonic, atonic, or autonomic phenomena. Patients usually have a previous diagnosis of generalized epilepsy. Absence status presents as a nonconvulsive seizure lasting between a half hour and several days. It usually ends spontaneously and suddenly but should be treated with antiseizure medications at the time of diagnosis.[16]
Evaluation
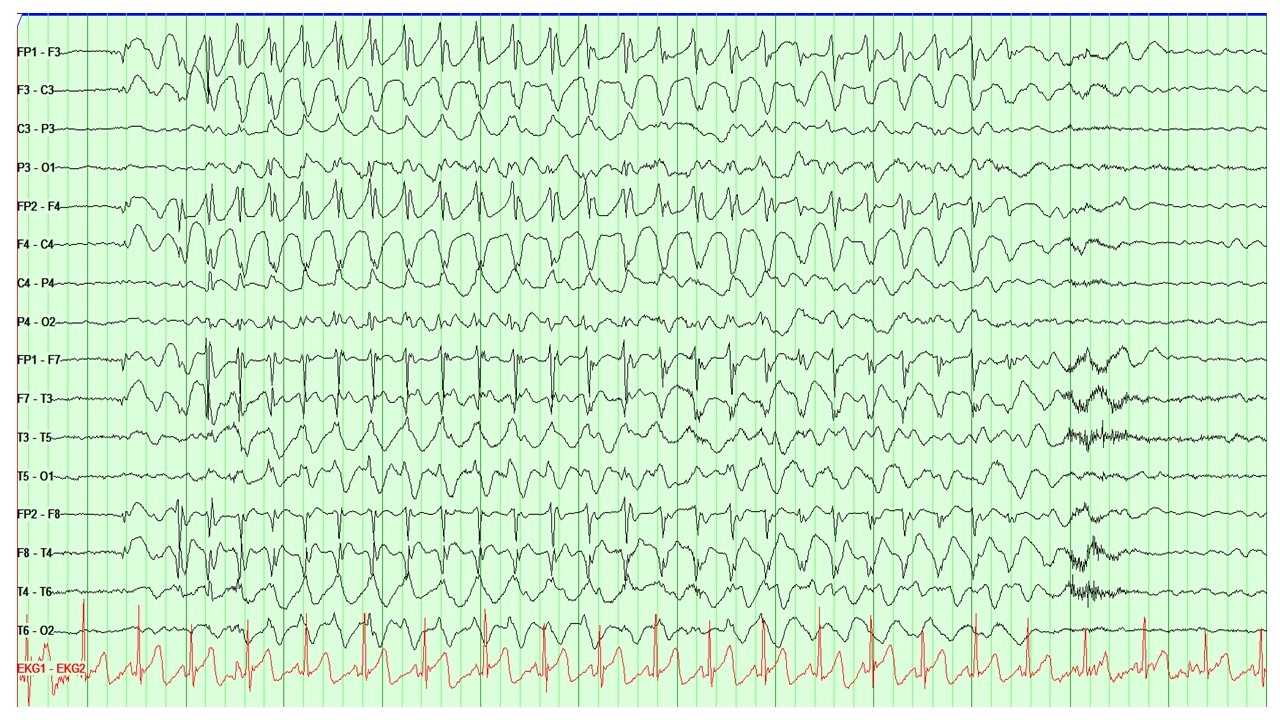
EEG is the main diagnostic tool for the evaluation of absence epilepsy. In the case of childhood absence seizures, EEG shows bilaterally synchronous and symmetrical 3-Hz spike and wave discharges that start and end abruptly (see Image. EEG Showing the Characteristic 3-Hz Spike and Wave Discharges Seen in Absence Epilepsy). These discharges sometimes have maximum frontal amplitude or begin with unilateral focal spikes.[2] In 50% of seizures in CAE, the initial discharge seen has a typical spike and wave morphology. The remaining 50% can show a single spike, polyspikes, or an atypical, irregular, generalized spike and wave on a normal background.[1][4]
Atypical absence seizures show a more insidious onset and offset, slower spike and wave paroxysms (slower than 3 Hz), and an abnormal interictal background.[4] In JAE, EEG demonstrates paroxysms of generalized 3- to 4-Hertz spike-and-wave or polyspike and wave discharges.[4]
Neuropsychological testing has shown that patients with CAE have a higher rate of cognitive deficits, especially involving attention, executive function, and verbal and visuospatial memory. Difficulty with language and reading is also commonly reported. Depression, anxiety, and attention-deficit/hyperactivity disorder also have been reported more frequently in patients with CAE.[1]
In the case of absence status, EEG shows continuous or nearly continuous generalized spike and wave or polyspike and wave discharges at 2 to 4 Hz. In patients with a new diagnosis of absence seizures, brain imaging is indicated to assess for structural abnormalities.
Treatment / Management
The first-line treatment for absence epilepsy is ethosuximide. A randomized controlled trial in 2010 that included 446 children with CAE showed that ethosuximide and valproic acid were superior to lamotrigine.[17] However, this study had a low seizure-free rate, with 53% of patients in the ethosuximide group, 58% in the valproic acid group, and 29% in patients taking lamotrigine. The group that received valproic acid had significantly lower scores on attentional measures compared to the ethosuximide and lamotrigine groups. For this reason, ethosuximide is the preferred agent for treating absence epilepsy. A study showed that only one-quarter of children with absence epilepsy became seizure-free with levetiracetam. If effective, levetiracetam can control absence epilepsy at a relatively low dose (usually <40 mg/kg/d). Increasing doses are not effective if levetiracetam is not initially effective.[18]
The most frequent adverse effects of ethosuximide are abdominal pain and nausea. For this reason, ethosuximide should be taken with meals. Other medications that can be used to manage CAE include valproate, lamotrigine, and topiramate. Second-line medications that can be used as adjunct therapy include valproic acid, zonisamide, and levetiracetam. It is important to note that some sodium channel blockers like phenytoin, carbamazepine, gabapentin, pregabalin, and vigabatrin can worsen absence seizures.[4]
Women of childbearing age not using contraception should not be treated with valproic acid; the preferred agent is ethosuximide.
Some experts suggest that a ketogenic or a medium-chain triglyceride diet may be beneficial, but strong evidence to support their use is lacking.
Differential Diagnosis
The differential diagnosis for staring spells includes absence epilepsy, focal seizures with alteration of awareness, and nonepileptic paroxysmal events.
Focal epilepsy with alteration of awareness (previously called complex partial epilepsy) can also present with behavioral arrest and automatisms. However, these seizures are usually less frequent than absence seizures. Patients can have generalized seizures with focal epilepsy. The semiology of automatisms can vary depending on the area of the cerebral cortex where the seizures originate.
A study that evaluated nonepileptic staring spells using video-EEG monitoring found that these episodes were often characterized by arrest of all activity, vague facial expressions, and vision fixed on 1 point without blinking. When the duration of the events was quantified, staring episodes lasted between 3 and 74 seconds. In most children, it was difficult to determine the onset and end of the event. Most parents could not regain the child’s attention by waving their hands in front of the child. Other more energetic measures like hand clapping or other loud sounds successfully stopped the events in all children. A significant percentage of children (41%) were inactive at the onset of the stare, and 18% were watching television when the event began.[19]
A retrospective chart review performed in a tertiary care epilepsy center showed that among 276 patients in the epilepsy monitoring unit, only 11% were deemed to have seizures.[20] Therefore, most staring spells are nonepileptic in nature. Clinicians should be careful when patients present with staring spells and not tell parents or other providers that these episodes are “absence seizures” before EEG evaluation is completed. The study mentioned above developed a tool to determine the pretest probability of seizures in children presenting with staring spells. This tool accounts for patient variables such as results of previous EEG, previous use of antiseizure medications or treatments for psychiatric conditions, and duration of the spells.[20]
Prognosis
Typical CAE occurs in childhood and resolves by adolescence. Seizure freedom is reported in 57% to 74% of the patients. Compared with controls, the risk for accidental injuries during absence seizures is well-reported.[19] As mentioned before, these patients have problems in the areas of attention, executive function, and verbal and visuospatial memory. Difficulty with language and reading is also commonly reported. Depression, anxiety, and ADHD have also been reported more frequently in patients with CAE.[1]
Complications
Some activities can be dangerous for people with absence seizures because of the temporary loss of awareness accompanying these episodes. Swimming, operating hazardous machinery, and driving during an absence seizure might result in drowning or an accident. Patients may need to restrict certain activities until their seizures are under control. Some states may also have laws regarding how long a patient must go without a seizure before resuming driving.
Consultations
A general pediatric neurologist or a pediatric epileptologist should be consulted when a patient has staring spells suspected to be seizures. Typically, an outpatient evaluation is a reasonable first step.
Deterrence and Patient Education
Most staring spells are nonepileptic in nature. When patients present with staring spells, clinicians should explain to caregivers that the differential diagnosis includes seizures, but they should avoid giving a specific diagnosis of “absence seizures” before an EEG evaluation is completed. Requesting caregivers to take videos of staring spells can be very useful, as this can help characterize them. Event calendars/logs can assist in understanding the frequency, pattern, and possible triggers.
Caregivers of children with CAE should know that generalized tonic-clonic seizures are rare. For this reason, rescue medications such as rectal diazepam and intranasal midazolam are not routinely prescribed. Nevertheless, caregivers should be taught what to do if the child has a generalized tonic-clonic seizure.
Caregivers often assume that absence seizures are harmless since they are very brief. Occasionally, they question the need to treat them with medications, arguing that the risks could outweigh the benefits. In these situations, it is recommended to explain that the child is experiencing frequent episodes of altered consciousness that can increase the risk of accidents. Seizures can also interfere with learning and negatively impact school performance. Activities like swimming, diving, or rock climbing should only be permitted under supervision. Driving is not permitted if seizures are uncontrolled and can vary by location.
Pearls and Other Issues
Clinical pearls for absence seizures offer valuable insights for clinicians navigating the complexities of this neurological condition.
- Absence epilepsy is classified as a typical or atypical absence, depending on seizure characteristics and EEG patterns.
- Absence seizures are characterized by behavioral arrest and EEG showing 3-Hz spike and wave discharges. Episodes usually occur multiple times per day.
- Absence seizures are seen in several generalized epilepsies, including childhood absence epilepsy (CAE), juvenile absence epilepsy (JAE), and juvenile myoclonic epilepsy (JME).
- Episodes of behavioral arrest should be called “staring spells” until an EEG evaluation is performed. Patients can only be diagnosed with absence epilepsy if they have characteristic seizure semiology and EEG findings.
- Staring spells can be epileptic or nonepileptic, with the majority being nonepileptic. When episodes are epileptic, they may be due to absence seizures or focal seizures with impaired awareness (complex partial seizures).
- The first-line treatment for absence seizures is ethosuximide. Other therapies include valproate, lamotrigine, and topiramate. Second-line medications that can be used as adjunct therapy include zonisamide and levetiracetam.
- Carbamazepine, phenytoin, gabapentin, vigabatrin, and other medications with similar mechanisms of action can worsen absence epilepsy.
- Approximately 60% of patients with CAE eventually achieve seizure freedom.
- Lack of response to ethosuximide and the presence of generalized tonic-clonic seizures have been associated with a lack of seizure remission and life-long epilepsy.
Enhancing Healthcare Team Outcomes
Neurologists are key to managing absence seizures. However, the long-term care and monitoring are done by an interprofessional team that includes the neurologist (child neurologist in the case of the pediatric population), primary care physician, advanced care practitioner, nurse, and pharmacist.
Physicians and advanced care practitioners must possess a nuanced understanding of absence seizure diagnosis and management, employing evidence-based practices with ethical considerations. The primary care practitioner, together with the neurologist, should follow up on the liver function tests, amylase, and complete blood count. In addition, birth control should be managed in women taking valproic acid. Finally, drug levels should be monitored periodically as these drugs have a narrow therapeutic index.
Nurses play a crucial role in managing absence seizures by closely monitoring patients for seizure activity, documenting symptoms, and promptly alerting the healthcare team to any changes. Additionally, they educate patients on medication adherence, seizure triggers, and safety precautions, empowering them and caregivers to effectively manage the condition outside clinical settings.
The pharmacist should educate the patient on the importance of medication compliance. In addition, the pharmacist should ask about any adverse reaction at the time of dispensing the medication. The caregiver should be asked if the symptoms are controlled; if not, the pharmacist should communicate with the interprofessional team about the need for a change in the agent used or dosage. A few patients with intractable absence epilepsy may be candidates for the ketogenic diet. If this is the case, a registered dietician with expertise in this diet should be consulted and periodically evaluate the patient. Mental health providers may be involved in the patient's care as many of these patients develop anxiety, depression, and stress. Therapists with expertise in holistic neurologic care are an important part of the healthcare team.
The interprofessional team care model represents the optimal approach to accurately diagnosing, treating, and supporting patients with this often-overlooked seizure disorder. Most patients need lifelong follow-up, and the interprofessional team must communicate with each other at any time when there is a change in drug dose, change in drug type, or development of adverse drug reactions. Optimizing the healthcare team's performance enhances clinical practice and improves outcomes for individuals living with absence seizures.