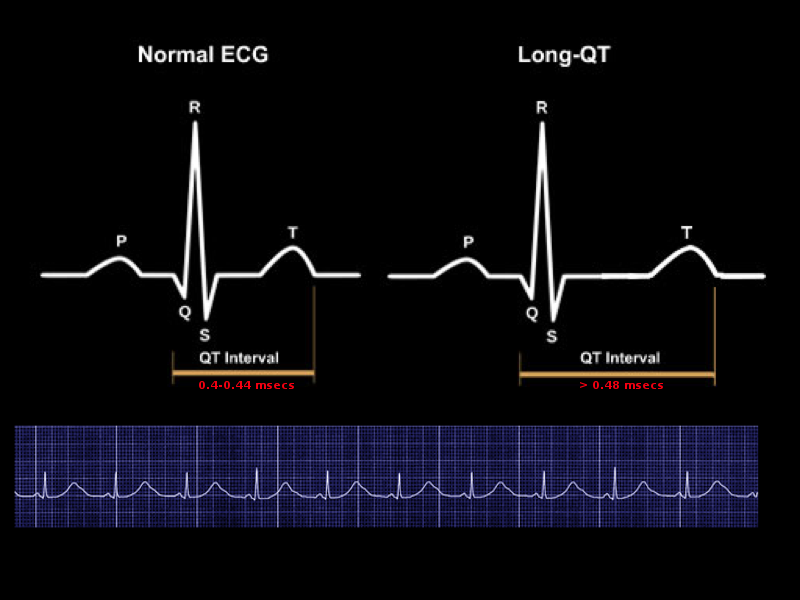
[1]
JERVELL A, LANGE-NIELSEN F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. American heart journal. 1957 Jul:54(1):59-68
[PubMed PMID: 13435203]
[2]
Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Tranebjærg L, Samson RA, Green GE. Jervell and Lange-Nielsen Syndrome. GeneReviews(®). 1993:():
[PubMed PMID: 20301579]
[3]
Uysal F, Turkgenc B, Toksoy G, Bostan OM, Evke E, Uyguner O, Yakicier C, Kayserili H, Cil E, Temel SG. "Homozygous, and compound heterozygous mutation in 3 Turkish family with Jervell and Lange-Nielsen syndrome: case reports". BMC medical genetics. 2017 Oct 16:18(1):114. doi: 10.1186/s12881-017-0474-8. Epub 2017 Oct 16
[PubMed PMID: 29037160]
Level 3 (low-level) evidence
[4]
Shih HT. Anatomy of the action potential in the heart. Texas Heart Institute journal. 1994:21(1):30-41
[PubMed PMID: 7514060]
[5]
Ackerman MJ, Priori SG, Dubin AM, Kowey P, Linker NJ, Slotwiner D, Triedman J, Van Hare GF, Gold MR. Beta-blocker therapy for long QT syndrome and catecholaminergic polymorphic ventricular tachycardia: Are all beta-blockers equivalent? Heart rhythm. 2017 Jan:14(1):e41-e44. doi: 10.1016/j.hrthm.2016.09.012. Epub 2016 Sep 19
[PubMed PMID: 27659101]
[6]
Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, Shkolnikova M, Berul CI, Bitner-Glindzicz M, Toivonen L, Horie M, Schulze-Bahr E, Denjoy I. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation. 2006 Feb 14:113(6):783-90
[PubMed PMID: 16461811]
Level 2 (mid-level) evidence
[7]
Früh A,Siem G,Holmström H,Døhlen G,Haugaa KH, The Jervell and Lange-Nielsen syndrome; atrial pacing combined with ß-blocker therapy, a favorable approach in young high-risk patients with long QT syndrome? Heart rhythm. 2016 Nov
[PubMed PMID: 27451284]
[8]
Green JD, Schuh MJ, Maddern BR, Haymond J, Helffrich RA. Cochlear implantation in Jervell and Lange-Nielsen syndrome. The Annals of otology, rhinology & laryngology. Supplement. 2000 Dec:185():27-8
[PubMed PMID: 11140992]
[9]
Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Alders M, Bikker H, Christiaans I. Long QT Syndrome. GeneReviews(®). 1993:():
[PubMed PMID: 20301308]
[10]
Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Napolitano C, Timothy KW, Bloise R, Priori SG. CACNA1C-Related Disorders. GeneReviews(®). 1993:():
[PubMed PMID: 20301577]
[11]
Nguyen HL, Pieper GH, Wilders R. Andersen-Tawil syndrome: clinical and molecular aspects. International journal of cardiology. 2013 Dec 5:170(1):1-16
[PubMed PMID: 24383070]
[12]
Kallergis EM, Goudis CA, Simantirakis EN, Kochiadakis GE, Vardas PE. Mechanisms, risk factors, and management of acquired long QT syndrome: a comprehensive review. TheScientificWorldJournal. 2012:2012():212178. doi: 10.1100/2012/212178. Epub 2012 Apr 19
[PubMed PMID: 22593664]
[13]
Goldenberg I, Moss AJ, Zareba W, McNitt S, Robinson JL, Qi M, Towbin JA, Ackerman MJ, Murphy L. Clinical course and risk stratification of patients affected with the Jervell and Lange-Nielsen syndrome. Journal of cardiovascular electrophysiology. 2006 Nov:17(11):1161-8
[PubMed PMID: 16911578]
Level 2 (mid-level) evidence