Continuing Education Activity
Pulmonary hamartomas are benign malformations of the lung and include an abnormal mixture of tissue components such as cartilage, epithelium, fat, or muscle which are common to the lung but the organ architecture is not preserved. This activity describes the evaluation and management of pulmonary hamartoma and explains the role of the interprofessional team in managing patients with this condition.
Objectives:
- Describe the epidemiology of pulmonary hamartoma.
- Identify the typical 'popcorn' or 'comma-shaped' appearance of calcification on imaging of pulmonary hamartoma.
- Outline the use of surgery in the management of pulmonary hamartoma.
- Review the importance of collaboration and communication among the interprofessional team members to improve the delivery of care for patients affected by pulmonary hamartoma.
Introduction
A lesion first described by German pathologist Eugen Albrecht in 1904, hamartomas are generally benign tumors that may occur in the lungs, skin, heart, breast and other regions of the body. The word hamartoma derives from “hamartia,” the Greek word for ‘erroneous,” or “faulty."[1] The cellular make-up of a hamartoma is that of an abnormal mixture of tissue components that are common in the organ of origin, though organ architecture is usually not preserved within the lesion.
Pulmonary hamartomas can, therefore, be viewed as benign malformations of common lung tissue, to include cartilage, epithelium, fat, or muscle. They are the most common benign pulmonary neoplasm in adults. However, they are much more rare in children.[2][3] These lesions are usually located in the peripheral parenchyma but have in rare cases been present in the central chest wall. Lesions tend to grow at the same rate as the surrounding tissue. Therefore neoplastic pressure erosion of adjacent structures is typically not noted.
Etiology
Historically, no specific risk factors have been identified for pulmonary hamartoma. Occasionally, a relationship between lesions that have undergone sarcomatous transformation and specific genomic alterations has been described,[4] but there are currently no screening guidelines specifically designed for the early diagnosis of pulmonary hamartoma.
Epidemiology
Pulmonary hamartomas occur with an incidence of 0.025 to 0.032% within the adult population. They usually present in the fifth and sixth decade of life, with males being four times more likely to be affected than females.[3] Although still an uncommon occurrence, these lesions are the most common benign pulmonary neoplasm, accounting for an estimated 77% of benign lung nodules and 8% of solitary lung lesions.[2][5] Most hamartomas occur in the peripheral parenchyma, with exceptions observed in the central chest wall. Additionally, approximately 10 % of lesions present endobronchially.[2][6] Within the pediatric population, pulmonary hamartomas are significantly rarer.[3]
Histopathology
Also known as chondroid hamartomas, the histological makeup of these tumors is a mixture of mature mesenchymal tissue, like adipose tissue, cartilage, bone, or smooth muscle bundles, and fibromyxoid tissue, with varying proportions of each component. They are non-invasive, slow-growing, nodular lesions, sometimes displaying cleft-like spaces within, lined by respiratory epithelium.[3]
History and Physical
In adults the majority of parenchymal hamartomas produce no symptoms, often being incidental findings. Depending on the location and size of the lesion, however, patients can still develop an array of complaints, including persistent coughing or wheezing, dyspnea, hemoptysis, rhonchi, higher likelihood of pneumonia, atelectasis or even pneumothorax. Endobronchial masses additionally pose the danger of airway obstruction. Hamartomas that have become symptomatic require a thorough diagnostic approach, and surgical resection can become necessary.[3][7]
Evaluation
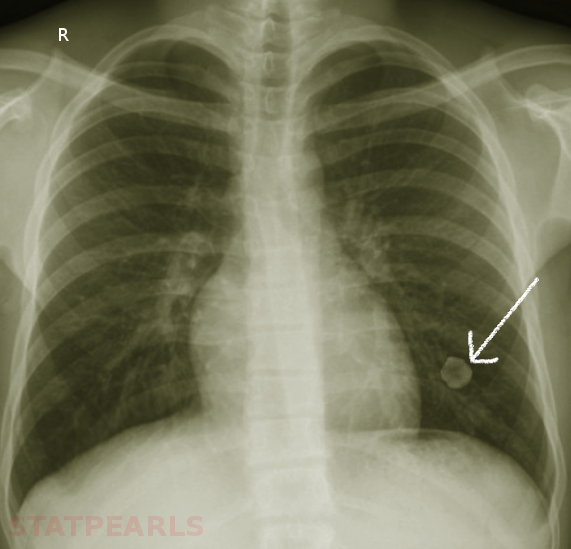
Hamartomas are often incidental findings on imaging and can mimic pulmonary malignancies. Once found, or in the case of asymptomatic hamartoma, there are multiple diagnostic strategies available to determine the nature of the lesion. On imaging, e.g., chest X-ray or CT scans, masses present as coin- shaped and solitary, with well-defined edges, typically measuring less than 4 cm in diameter. Calcification is present in 25% to 30% of patients. The “popcorn” or “comma-shaped” appearance of calcification is pathognomonic for hamartomas.[8][5] While CT imaging remains the gold-standard, further diagnostic measures can become necessary. FDG-PET scan can be useful to determine the rate of FDG uptake, and therefore metabolic rate, of lesions with an indeterminate risk of malignancy.[5] Especially in the case of lesions with an absent adipose component, or those lacking the characteristic calcification pattern, biopsy becomes mandatory to rule out underlying malignancy. Bronchoscopy with fine needle aspiration (FNA) is commonly the strategy of choice, though aspirations can be scant due to the density of the lesions. Masses with the typical coin appearance that fulfill CT criteria for hamartoma (less than 4cm in size, well-defined edges, detectable calcification or fatty component) should remain subject to conservative follow up with periodical observation. Resection is reserved for fast-growing or symptomatic masses or those in which the possibility of malignancy cannot be excluded.[9][10]
Treatment / Management
Surgery remains the only definitive curative option available. In the event of surgery, preservation of functional lung tissue is the primary goal. Therefore, enucleation and wedge resections are the most common surgical choice, with more radical lobectomy or total pneumonectomy reserved for particularly deep lesions, multiple or large lesions that are making wedge resection impossible, or lesions adhering severely to the hilum of the lung. To avoid overlooking underlying malignant potential, obtaining intraoperative frozen sections is generally recommended.[11]
Differential Diagnosis
The most significant consideration when encountering a possible hamartomatous lesion should go to distinguishing the lesion from and excluding the possibility of underlying malignancy. Upon exclusion of malignancy, other benign pulmonary tumors should be considered when deliberating the nature of a solitary pulmonary nodule, to include infectious granuloma, lipoma, lipoid pneumonia, or pulmonary papilloma.[12] Also, though 90% are solitary occurrences, pulmonary hamartomas can also appear in association with genetic syndromes.[5] Multiple pulmonary chondromatous hamartomas have been noted as manifestations of either the Carney triad or Cowden syndrome. The former is predominantly seen in young women and characterized by the concurrent appearance of gastric leiomyoblastoma, pulmonary hamartoma, and extra-adrenal paraganglioma. Patients with Cowden disease often display multiple hamartomas, manifesting as mucocutaneous lesions, multiple benign tumors of internal organs and an increased risk for several forms of cancer, including breast and digestive tract malignancies.[13][14]
Prognosis
The prognosis for patients with lung hamartoma is generally excellent. Lesions are slow-growing and, in cases where symptoms are present and persistent, surgery is curative. Malignant transformation or subsequent malignancy are rare occurrences, and if patients adhere to a conservative observation schedule, are likely to be diagnosed early on.[3][11]
Complications
Beyond the possibility of airway obstruction, with subsequent atelectasis or recurrent pneumonia, pulmonary hamartomas have in rare cases been noted to bear potential for sarcomatous transformation. Common signs of malignant alteration are the rapid growth of the lesion and systemic symptoms like weight loss, weakness or fatigue. Histologically, this malignant change may not always be apparent, however macroscopic signs, like an invasion of adjacent tissue or distant metastases may be present. In some cases, a possible relationship between the presence of genomic abnormalities of 12q14 and 6q21, encoding the high mobility group A (HMGA), and the occurrence of pulmonary hamartoma has been observed. Also, incidence rates of lung cancer have been reported to be six times higher in patients with pulmonary hamartomas than in healthy subjects, making genetic predisposition to either malignant change or subsequent development of malignancy a possibility.[4][15]
Deterrence and Patient Education
There are no risk factors associated with the development of pulmonary hamartomas, nor are there defined screening guidelines, considering the mostly sporadic nature of the lesions. Considering the risk, albeit small, of malignant change and development of subsequent malignancy, patients should be advised to adhere to a schedule conservative observation in the form of periodical imaging and comparison with previous results.[11][4][15]
Enhancing Healthcare Team Outcomes
Patients with lung lesions often first come to the attention of the primary care provider and nurse practitioner. Because there is always the possibility of malignancy, these patients should obtain a referral to an interprofessional team that includes an oncologist, thoracic surgeon, radiologist, and a pulmonologist. If observation is deemed to be the management plan, the most important facet is to ensure that the lesion is not malignant. No matter how technically skilled the radiologist is interpreting lung lesions, the definitive diagnosis is only possible with a biopsy. If observation is the chosen course, serial CT scans are recommended to ensure that the lesion is not growing.