Continuing Education Activity
Delayed puberty not infrequently occurs in the pediatric population and a common reason for referral to a pediatric endocrinologist. While it is usually a benign diagnosis, more serious causes should be considered and ruled out when warranted by clinical suspicion. This activity reviews the evaluation and management of pubertal delay and highlights the role of interprofessional team members in collaborating to provide well-coordinated care to patients with this condition.
Objectives:
- Identify the etiology of delayed puberty.
- Recall the history, physical, and evaluation of delayed puberty.
- List the treatment and management options available for delayed puberty.
- Review the importance of improving care coordination amongst interprofessional team members to improve outcomes for patients affected by delayed puberty.
Introduction
Puberty can be a stressful and concerning time for adolescents and their families, as it represents a period of significant emotional and physical changes to the body. Puberty normally occurs between the ages of 8 to 13 years for females and 9 to 14 years for males. In females, the first sign of true puberty is breast development or thelarche. In males, the first sign is testicular enlargement, where testicular size increases to a volume of 4 ml or greater, or the length of 2.5 cm. When variation exists on the timing and onset of puberty, the primary pediatrician or provider must closely monitor the child's development and ensure that any pathological or reversible causes are ruled out.
When a child exhibits early signs of puberty, it is defined as precocious puberty. This condition occurs when the first signs of puberty appear before 8 years in females and 9 years in males. However, this activity discusses delayed puberty for females and males. In females, delayed puberty is the lack of breast development by 13 years, a delay of over 4 years between thelarche and completion of puberty, or a lack of menarche by 16 years. In males, a pubertal delay is evident by a lack of testicular enlargement by 14 years or more than 5 years between testicular enlargement and completion of puberty.[1] Puberty represents the maturation of the hypothalamic-pituitary-gonadal (HPG) axis. Development of acne, axillary or pubertal hair, and body odor are a result of adrenal androgen secretion and defined as adrenarche[2] Adrenarche is independent of the HPG axis. Therefore, a child could display signs of adrenarche but still have a diagnosis of pubertal delay.[3]
Etiology
Delayed Puberty in Males and Females
Common causes of delayed puberty for both males and females can be from functional hypogonadotropic hypogonadism - this is normally a temporary clinical state brought on by different stresses to the body, including chronic illnesses such as severe persistent asthma, sickle cell anemia, cystic fibrosis, or ulcerative colitis. Any cause of nutritional deficiency also merits consideration. While eating disorders occur more in females, it would be wrong to dismiss them in males. The provider should also investigate any external causes for malnutrition, including the patient's social situation at home. In developing adolescents, psychological causes must also be investigated and frequently correlate with the above conditions mentioned. A rarer cause of hypogonadotropic hypogonadism that is congenital is panhypopituitarism. However, this typically results in growth hormone deficiency as well and should be considered if a patient presented early in age with severe short stature.[3]
Delayed Puberty in Males
In males, a common cause of pubertal delay is a constitutional delay of puberty and growth (CDPG). CDPG occurs when there is a decrease in the tempo of growth. The patients are generally healthy adolescent males, who appear short for age. At birth, they are an average size. However, their rate of growth slows down around 3 to 6 months of age. When they reach 3 or 4 years of age, the patients will be growing below but parallel to the 3rd percentile line. As their male peers experience puberty and a growth spurt, the patient will continue to have a lower growth velocity (2 to 4 cm/year) and pubertal delay. In normal boys, the growth spurt occurs at a testicular volume of 10 ml, which is between Tanner stage 3 and 4 (usually ages 13 to 15). In boys with CDPG and delayed puberty, their growth spurt occurs at a later age, generally between 15 to 17 years of age.[4] When the patient finally experiences puberty, his catch-up growth will continue until he reaches his predicted target height, which may not occur until he is older than 17 or 18 years. The patient's bone age will experience delay compared to his chronological age by 2 or more years; the bone age will also correlate with his current height.[3] History will typically reveal a sibling or parent that was a "late bloomer." For example, the father may not have experienced a growth spurt until 15 or 16 years of age. CDPG is normally a diagnosis of exclusion. However, it is often difficult to differentiate between CDPG and hypogonadotropic hypogonadism.
Hypogonadotropic hypogonadism occurs when there is a permanent delay in the maturation of the HPG axis. There is the partial or complete deficiency of gonadotropin-releasing hormone (GnRH), causing decreased luteinizing hormone (LH) and follicle-stimulating hormone (FSH) release, which ultimately leads to decreased testosterone production.[5] Hypogonadotropic hypogonadism may be idiopathic or congenital. If the patient has anosmia, or the absence of smell, Kallman syndrome (KS) merits strong diagnostic consideration. Kallman syndrome results from a genetic mutation of the KAL1 or FGFR1 (fibroblast growth factor receptor 1) genes.[6] The development of the olfactory system has a tight connection with the migration of GnRH neurons during early embryogenesis. When an issue occurs with GnRH migration, the olfactory system becomes negatively impacted, leading to the loss of smell. Other associated physical findings may include cleft lip, cleft palate, hypodontia, eye defects, or hearing impairments. A brain MRI may help to support the diagnosis or rule out the presence of a tumor or lesion along the HPG axis. Brain tumors such as adenomas and craniopharyngiomas are rare causes of hypogonadotropic hypogonadism, but they are a more common cause of pubertal delay in males than in females. Such masses disrupt the HPG axis, causing a downstream decrease in sex hormones. Suspicion for a cranial mass is advisable when a child presents with headaches, dizziness, vomiting, and changes in vision.
On the other hand, hypergonadotropic hypogonadism results when there is a primary gonadal failure. Testosterone levels are low, causing an increase in GnRH, LH, and FSH. The etiology of hypergonadotrophic hypogonadism can be either acquired or congenital. Acquired causes include radiation to testes for malignancy, surgery for cryptorchism or torsion, or an infection such as orchitis from mumps.[3] In males, the most common congenital form of primary gonadal insufficiency is Klinefelter syndrome. Klinefelter syndrome is due to aneuploidy of the sex chromosomes, most commonly resulting in a 47, XXY karyotype. Patients present with tall stature, disproportionately long limbs, eunuchoid body habitus, gynecomastia, and neurological or behavioral problems. However, the hallmark sign is small (less than 4 ml) but firm testes; this usually leads to infertility due to oligospermia or azoospermia.[7] Lastly, patients with hypergonadotropic hypogonadism could have vanishing testis syndrome, also known as testicular regression syndrome (TRS). TRS occurs in 5% of cryptorchism cases. While the cause of TRS is still unclear, the hypothesis is that an event of vascular thrombosis or torsion, occurring in the antenatal or perinatal period, causes testicular degeneration. Therefore, a fetus who initially developed normal testes in utero will be born with non-palpable testes and a rudimentary spermatic cord.[8]
Delayed Puberty in Females
Constitutional delay in puberty and growth is less common in females. When it does occur, family history normally includes a sibling or parent who was a "late bloomer." However, functional hypogonadotropic hypogonadism is much more common in females. It usually develops secondary to conditions that reduce total body fat, which is commonly associated with anorexia nervosa or excessive exercise in females. Both involve a significant reduction in calories that also decreases the body's leptin concentration, resulting in gonadotropin deficiency. Decreased LH and FSH secretion combined with lower body fat depresses estrogen production and secretion, thus delaying puberty. Kallman syndrome can also be a cause for delayed puberty in females, but it is very rare and more common in males. This predilection is because KS is an X-linked recessive genetic disorder, but can also be autosomal dominant.[5]
In females, hypergonadotropic hypogonadism results from primary ovarian failure and is either acquired or congenital. Acquired causes include receiving radiation therapy for the treatment of cancers and malignancies. Autoimmune destruction of the ovaries could also lead to hypergonadotrophic hypogonadism. However, this is usually associated with other autoimmune disorders in patients who have more than one autoimmune diagnosis like type I diabetes mellitus or Hashimoto thyroiditis (autoimmune polyglandular type I or type II syndromes).[3] When hypergonadotropic hypogonadism is associated with short stature, Turner syndrome (TS) must be a consideration. Turner syndrome results from a partial or complete absence of an X chromosome, producing the 45, X karyotype. Patients may present in infancy with nuchal translucency, cystic hygroma, or lymphedema. Common clinical features also include a webbed neck, broad chest with widely spaced nipples, and short stature.[9] Other common associations with TS include a bicuspid aortic valve, coarctation of the aorta, autoimmune disorders like celiac disease, and congenital kidney malformations like horseshoe kidneys. Therefore, the management of TS may require an interprofessional team of different specialists.[10]
Epidemiology
In a large retrospective study, with 232 subjects at an academic center in the United States, the frequency of delayed puberty was divided by its different causes.[11]
- The most common cause of delayed puberty was CDPG, affecting 53% of adolescents 18 years or younger. CDPG was more common in males (63%) than in females (30%).
- Functional hypogonadotropic hypogonadism occurred in 19% of patients.
- Permanent hypogonadotropic hypogonadism comprised 12% of patients.
- Primary gonadal failure occurred in 13% of patients.
- Patients without a clearly classified disorder occurred in 3% of subjects.
History and Physical
When patients and their families come with a concern for the pubertal delay, obtaining a good history is essential to a thorough evaluation.
- History of Present Illness: The provider should first inquire about any signs of puberty the child or the caregivers have noticed, such as breast development, testicular enlargement, body odor, axillary hair, pubertal hair, or acne. The occurrence of adrenarche versus puberty should be distinguished. A thorough review of systems from head to toe will help to rule in or rule out the causes of pubertal delay. Fatigue or weight loss could be concerning for a chronic condition such as sickle cell anemia, depression, or malnutrition. A young child complaining of headaches and blurry vision should undergo evaluation for a brain mass.
- Medical History: Birth history, immunization status, and the involvement of other specialties in the child's care are all pertinent to medical history. Does the patient have any medical conditions like asthma or cystic fibrosis?
- Medications/Treatments: If the patient had a malignancy in the past, did they receive any total body radiation for treatment?
- Family History: Were any biological siblings or parents considered "late bloomers" in their family?
- Surgical History: Did the patient have any previous surgical correction of cryptorchidism, which would result in primary gonadal failure?
- Social history: How is the patient's home environment? Do they live with both parents? Questions about having concerning behavior or mood instability should also merit investigation.
- Development: Has the patient missed any milestones or been diagnosed with any developmental delay? Disorders such as Klinefelter syndrome is commonly associated with developmental delay or behavioral issues.
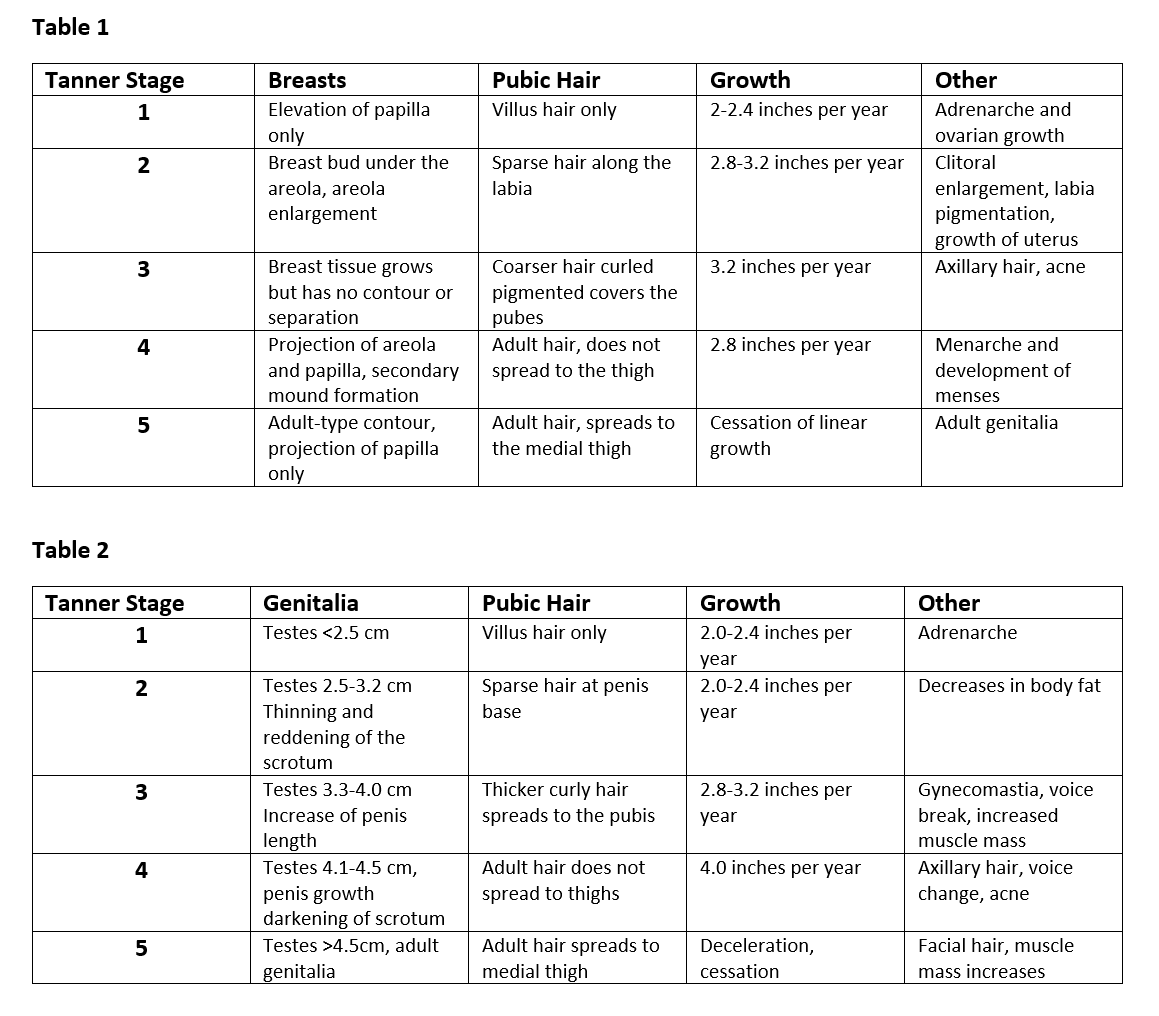
A complete physical exam should always be done to deny or support any clinical suspicions. Before examining the child, assess vital signs, including height, weight, and body mass index (BMI). Using previous or current growth curves that are appropriate for age and sex will help portray any concerning patterns. Note any signs of adrenarche on examination, including body odor or facial acne. Lastly, assigning a Tanner stage must always be included. Please see the below figure for appropriate Tanner staging.
Evaluation
Before ordering labs and imaging, the child's predicted target height, based on the biological parents' adult heights, should be plotted. It is essential to look over the patient's growth curves, based on appropriate age, sex, height, weight, and BMI. If multiple data points are available on the curves, any changes in pattern or velocity may be of significance.
If further workup is warranted, a bone age can help determine the child's current growth status. An X-ray of the left wrist and hand would evaluate for bone age. An abdominal ultrasound to look at the ovaries and uterus may help evaluate a patient with Turner syndrome looking for streak gonads. An ultrasound of the testicles can help with investigating cryptorchidism or a mass. When there is suspicion for a brain mass like a craniopharyngioma, the clinician should order a brain MRI.[12]
A provider may also choose to add laboratory tests to evaluate a child's pubertal status. Common tests to include are serum LH, FSH, estradiol in females, and total testosterone in males. Ordering additional labs should be lead by clinical findings and frequently includes a complete blood count, complete metabolic panel, free T4 and thyroid-stimulating hormone for thyroid function, and erythrocyte sedimentation rate for chronic inflammatory conditions. If a patient has galactorrhea with delayed puberty, an evaluation for a prolactinoma with a prolactin level are necessary. Some clinicians may add tests to access for adrenarche and may include DHEA-S. If panhypopituitarism or growth hormone deficiency is a concern, insulin-like growth factor 1 (IGF-1) require testing.[12][13] A pediatric endocrinologist may choose to perform a GnRH stimulation test in the future, but this is usually not included in the initial evaluation for delayed puberty. Lastly, if there is any suspicion for a syndrome, testing for karyotype should be considered.[3]
Treatment / Management
Constitutional Delay in Puberty and Growth
After making a diagnosis of CDPG, treatment is usually guided by patient and parental goals. Close observation can be appropriate, especially if puberty has started and adulthood stature is not of primary concern. A short-course treatment with low doses of testosterone for males or estrogen for females is often initiated when puberty and growth are true psychosocial causes of stress and low self-esteem for a child. History of bullying, poor academic performance, or dropping out of athletics may lead the provider to start treatment. Treatment can improve growth velocity, sexual maturation, and mental well-being without producing serious side effects, or significantly affecting the final target height.[14] For males with CDPD, an oral and intramuscular (IM) form of testosterone are available. However, the IM form is usually more appropriate due to the serious side effect of liver toxicity caused by oral testosterone. In females with CDPG, an oral and IM form of estrogen are also available, but oral estrogen is more often the therapeutic choice. Once treatment has started, the patient must be monitored frequently for signs of pubertal development, which includes testicular enlargement in males or breast development in females. After a few months of therapy, the provider may choose to stop sex-steroid treatment to evaluate for continued pubertal development while off therapy.[3] For patients that are more concerned with short stature than with delayed puberty, growth hormone therapy has been used to augment height. However, it has not been shown to truly impact final adult height in adolescents with CDPG, and is thus, not recommended for treatment.[14]
Permanent Hypogonadism
Patients diagnosed with permanent hypogonadism, either from primary gonadal failure or permanent lesions in the HPG axis, will require a more prolonged course of sex-steroid therapy. In males, IM testosterone is the initial treatment of choice. A low dose of testosterone is started and increased gradually over time until achieving adult levels of testosterone. In females, a low dose of oral estrogen is the preferred initial treatment of choice. Estrogen is also increased incrementally over time until breakthrough vaginal bleeding occurs, or 12 to 24 months of treatment have passed. The recommendation is then for patients to start on combination estrogen and progesterone therapy for normal monthly withdrawal bleeding. The transition helps the body to experience more normal physiological menstrual cycles.[3] Commonly used hormone replacement therapies include oral contraceptives or transdermal estrogen patches with oral progesterone.
Differential Diagnosis
Delayed Puberty in Males and Females
- Chronic illnesses: sickle cell anemia, inflammatory bowel disease, cystic fibrosis, celiac disease, etc.
- Psychological: depression, anxiety
- Social: poor environment at home
Delayed Puberty in Males
- Constitutional delay of puberty and growth
- Hypogonadotropic hypogonadism
- Acquired
- Chronic illness: cystic fibrosis, sickle cell anemia, celiac disease, etc.
- Psychosocial: anxiety, depression
- Genetic
- Kallman syndrome
- Brain mass or tumor
- Hypergonadotropic hypogonadism
- Acquired
- Radiation therapy
- Testicular surgery
- Genetic
- Klinefelter syndrome
- Testicular regression syndrome
Delayed Puberty in Females
- Constitutional delay in puberty and growth
- Hypogonadotropic hypogonadism
- Acquired
- Chronic illness: cystic fibrosis, sickle cell anemia, celiac disease, etc.
- Psychosocial: anorexia nervosa, excessive exercise, depression, anxiety
- Genetic:
- Kallman syndrome
- Brain mass or tumor
- Hypergonadotropic hypogonadism
- Acquired
- Radiation therapy
- Surgery to ovaries
- Genetic
- Autoimmune ovarian failure
- Turner syndrome
Prognosis
The prognosis of delayed puberty depends on the underlying condition. CPDG generally has a good prognosis with either expectant therapy or treatment. The more complicated causes of pubertal delay may require additional specialists for support and care. Access to care and resources can also impact the patient's care and management if his or her diagnosis is complicated. Therefore, the prognosis is also dependent on an individual's clinical status and his or her financial and social support.
Complications
Pubertal delay can have many psychosocial influences during development and can cause emotional, social, or academic stress. In CDPG, it is essential to emphasize that this is simply a normal variation in pubertal timing and that treatment will not significantly impact predicted target height.[4] If the patient has other causes besides CDPG for the pubertal delay, it is vital that the provider evaluates the patient thoroughly so that there is a definitive diagnosis sooner, and management can commence earlier. Complications for the other causes of pubertal delay, excluding CDPG, depend on the underlying condition and differs accordingly.
Consultations
When a pubertal delay is concerning, a pediatric endocrinologist should provide a consult for further evaluation and management. The endocrinologist may choose to add additional imaging or laboratory tests, and can monitor the patient regularly for the initiation of therapy and follow up. If the patient's diagnosis is with something more complicated such as a brain tumor, then more specialists, including a neurologist and neurosurgeon, will be necessary.
Deterrence and Patient Education
Patients and families should receive education about what the first signs of puberty consist of in males and females. Counsel is also necessary about the normal timing of puberty, which is between 8 to 13 years in females and 9 to 14 years in males. When there is concern about early or delayed puberty, patients and caregivers should seek help from a medical provider for a prompt and accurate diagnosis. The primary provider may also choose to consult a pediatric endocrinologist, who may further evaluate and manage the patient.
Enhancing Healthcare Team Outcomes
Delayed puberty has repercussions beyond just the secondary sexual characteristics. It affects emotions, mood, behavior, social, and academic performance. Thus, the condition is best managed by an interprofessional team that deals with not only growth but the psychosocial aspect of the disorder.
Delayed puberty is often first recognized by the child's primary care provider. Once delayed puberty is suspected, further evaluation and management by a pediatric endocrinologist usually follows. The endocrinologist may order additional labs or imaging to investigate the cause for pubertal delay further; this involves the work and care of phlebotomists, laboratory technicians, radiology technicians, and radiologists.
If the underlying cause is more complicated, more specialists may need to be involved. For example, a malignant brain mass will necessitate the management of a neurologist, neurosurgeon, MRI technician, nurses, and pharmacists. At this point, the condition is more an oncology issue than a delayed puberty issue, and all these various providers will function in that capacity, which is beyond the purview of this activity.
Patients with poor self-esteem, depression, and lack of confidence should obtain a referral to a mental health nurse for counseling, who should report back to the health team regarding progress. Psychosocial treatment would potentially include pharmaceutical treatment for depression, and the pharmacist should weigh in since many psychologically active therapies carry additional risks in the adolescent population. The school nurse should provide support to help the child integrate with others. Parents require education about the disorder and its prognosis. The social worker should be involved to ensure that the patient has adequate social and financial support. Again, interprofessional collaboration and communication are crucial to achieving optimal patient outcomes, irrespective of the underlying cause. In particular, the nurse working with the patient and family must communicate with the clinical team to assure that appropriate family education is completed. Once therapy is initiated, monitoring becomes important and the nurse or primary care clinician must follow up with the specialists if milestones are not being reached. [Level V]
The outcomes for delayed puberty depend on its cause. However, to improve outcomes, prompt consultation with an interprofessional group of specialists is always recommended. Access to care and resources can also impact the patient's care and management if his or her diagnosis is complicated. Therefore, the prognosis is also dependent on an individual's clinical status and his or her financial and social support.