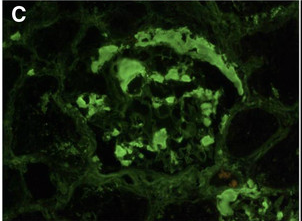
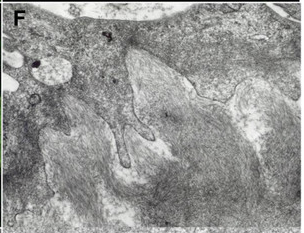
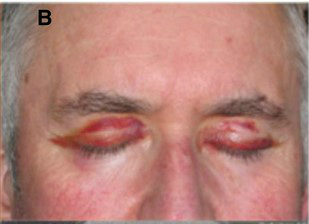
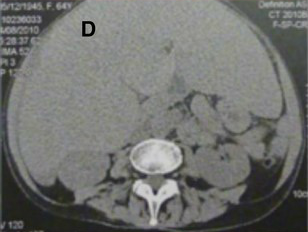
[1]
Tini G, Vianello PF, Gemelli C, Grandis M, Canepa M. Amyloid Cardiomyopathy in the Rare Transthyretin Tyr78Phe Mutation. Journal of cardiovascular translational research. 2019 Dec:12(6):514-516. doi: 10.1007/s12265-018-9859-0. Epub 2019 Jan 2
[PubMed PMID: 30604309]
[2]
Baiardi S, Rossi M, Capellari S, Parchi P. Recent advances in the histo-molecular pathology of human prion disease. Brain pathology (Zurich, Switzerland). 2019 Mar:29(2):278-300. doi: 10.1111/bpa.12695. Epub 2019 Jan 22
[PubMed PMID: 30588685]
Level 3 (low-level) evidence
[3]
Gavriatopoulou M, Fotiou D, Ntanasis-Stathopoulos I, Kastritis E, Terpos E, Dimopoulos MA. How I treat elderly patients with plasma cell dyscrasias. Aging. 2018 Dec 18:10(12):4248-4268. doi: 10.18632/aging.101707. Epub
[PubMed PMID: 30568029]
[4]
Thomas VE, Smith J, Benson MD, Dasgupta NR. Amyloidosis: diagnosis and new therapies for a misunderstood and misdiagnosed disease. Neurodegenerative disease management. 2019 Dec:9(6):289-299. doi: 10.2217/nmt-2019-0020. Epub 2019 Nov 5
[PubMed PMID: 31686587]
[5]
Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, Westermark P, Nomenclature Committee of the International Society of Amyloidosis. Amyloid fibril protein nomenclature: 2012 recommendations from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis. 2012 Dec:19(4):167-70. doi: 10.3109/13506129.2012.734345. Epub 2012 Nov 1
[PubMed PMID: 23113696]
[6]
Yakupova EI, Bobyleva LG, Vikhlyantsev IM, Bobylev AG. Congo Red and amyloids: history and relationship. Bioscience reports. 2019 Jan 31:39(1):. pii: BSR20181415. doi: 10.1042/BSR20181415. Epub 2019 Jan 15
[PubMed PMID: 30567726]
[7]
Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Sekijima Y. Hereditary Transthyretin Amyloidosis. GeneReviews(®). 1993:():
[PubMed PMID: 20301373]
[8]
Buck FS, Koss MN, Sherrod AE, Wu A, Takahashi M. Ethnic distribution of amyloidosis: an autopsy study. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 1989 Jul:2(4):372-7
[PubMed PMID: 2668942]
[9]
Alwahaibi NY, Al Issaei HK, Al Dhahli BS. Spectrum of glomerular diseases in Arab countries: A systematic review. Saudi journal of kidney diseases and transplantation : an official publication of the Saudi Center for Organ Transplantation, Saudi Arabia. 2018 Nov-Dec:29(6):1256-1266. doi: 10.4103/1319-2442.248285. Epub
[PubMed PMID: 30588955]
Level 1 (high-level) evidence
[10]
Andreou S, Panayiotou E, Michailidou K, Pirpa P, Hadjisavvas A, El Salloukh A, Barnes D, Antoniou A, Agathangelou P, Papastavrou K, Christodoulou K, Tanteles GA, Kyriakides T. Epidemiology of ATTRV30M neuropathy in Cyprus and the modifier effect of complement C1q on the age of disease onset. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis. 2018 Dec:25(4):220-226. doi: 10.1080/13506129.2018.1534731. Epub 2018 Dec 20
[PubMed PMID: 30572722]
[11]
Buxbaum JN. The systemic amyloidoses. Current opinion in rheumatology. 2004 Jan:16(1):67-75
[PubMed PMID: 14673392]
Level 3 (low-level) evidence
[12]
Mahmood S, Palladini G, Sanchorawala V, Wechalekar A. Update on treatment of light chain amyloidosis. Haematologica. 2014 Feb:99(2):209-21. doi: 10.3324/haematol.2013.087619. Epub
[PubMed PMID: 24497558]
[13]
Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011 May 10:29(14):1924-33. doi: 10.1200/JCO.2010.32.2271. Epub 2011 Apr 11
[PubMed PMID: 21483018]
[14]
Chuang E, Hori AM, Hesketh CD, Shorter J. Amyloid assembly and disassembly. Journal of cell science. 2018 Apr 13:131(8):. doi: 10.1242/jcs.189928. Epub 2018 Apr 13
[PubMed PMID: 29654159]
[15]
Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation. 2017 Apr 4:135(14):1357-1377. doi: 10.1161/CIRCULATIONAHA.116.024438. Epub
[PubMed PMID: 28373528]
[16]
Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, Wechalekar A, Gibbs SD, Venner CP, Wassef N, McCarthy CA, Gilbertson JA, Rowczenio D, Hawkins PN, Gillmore JD, Lachmann HJ. Senile systemic amyloidosis: clinical features at presentation and outcome. Journal of the American Heart Association. 2013 Apr 22:2(2):e000098. doi: 10.1161/JAHA.113.000098. Epub 2013 Apr 22
[PubMed PMID: 23608605]
[17]
McCausland KL, White MK, Guthrie SD, Quock T, Finkel M, Lousada I, Bayliss MS. Light Chain (AL) Amyloidosis: The Journey to Diagnosis. The patient. 2018 Apr:11(2):207-216. doi: 10.1007/s40271-017-0273-5. Epub
[PubMed PMID: 28808991]
[18]
Kapoor M, Rossor AM, Jaunmuktane Z, Lunn MPT, Reilly MM. Diagnosis of amyloid neuropathy. Practical neurology. 2019 Jun:19(3):250-258. doi: 10.1136/practneurol-2018-002098. Epub 2018 Dec 30
[PubMed PMID: 30598431]
[19]
Koop AH, Mousa OY, Wang MH. Clinical and endoscopic manifestations of gastrointestinal amyloidosis: a case series. Clujul medical (1957). 2018 Oct:91(4):469-473. doi: 10.15386/cjmed-951. Epub 2018 Oct 30
[PubMed PMID: 30564026]
Level 2 (mid-level) evidence
[20]
Vrana JA, Theis JD, Dasari S, Mereuta OM, Dispenzieri A, Zeldenrust SR, Gertz MA, Kurtin PJ, Grogg KL, Dogan A. Clinical diagnosis and typing of systemic amyloidosis in subcutaneous fat aspirates by mass spectrometry-based proteomics. Haematologica. 2014 Jul:99(7):1239-47. doi: 10.3324/haematol.2013.102764. Epub 2014 Apr 18
[PubMed PMID: 24747948]
[21]
Lisenko K, Schönland SO, Jauch A, Andrulis M, Röcken C, Ho AD, Goldschmidt H, Hegenbart U, Hundemer M. Flow cytometry-based characterization of underlying clonal B and plasma cells in patients with light chain amyloidosis. Cancer medicine. 2016 Jul:5(7):1464-72. doi: 10.1002/cam4.725. Epub 2016 Apr 25
[PubMed PMID: 27109862]
[22]
Westerland OA, Pratt G, Kazmi M, El-Najjar I, Streetly M, Yong K, Morris M, Mehan R, Sambrook M, Hall-Craggs M, Silver D, Goh V. National survey of imaging practice for suspected or confirmed plasma cell malignancies. The British journal of radiology. 2018 Dec:91(1092):20180462. doi: 10.1259/bjr.20180462. Epub 2018 Nov 1
[PubMed PMID: 30102561]
Level 3 (low-level) evidence
[23]
Kalle A, Gudipati A, Raju SB, Kalidindi K, Guditi S, Taduri G, Uppin MS. Revisiting renal amyloidosis with clinicopathological characteristics, grading, and scoring: A single-institutional experience. Journal of laboratory physicians. 2018 Apr-Jun:10(2):226-231. doi: 10.4103/JLP.JLP_148_17. Epub
[PubMed PMID: 29692592]
[24]
Eddou H, Zinebi A, Maaroufi HE, Moudden MK, Doghmi K, Mikdame M, Baaj ME. [Treatment of systemic AL amyloidosis: about 25 cases]. The Pan African medical journal. 2017:28():160. doi: 10.11604/pamj.2017.28.160.11885. Epub 2017 Oct 19
[PubMed PMID: 29541306]
Level 3 (low-level) evidence
[25]
Popkova T, Hajek R, Jelinek T. Monoclonal antibodies in the treatment of AL amyloidosis: co-targetting the plasma cell clone and amyloid deposits. British journal of haematology. 2020 Apr:189(2):228-238. doi: 10.1111/bjh.16436. Epub 2020 Feb 18
[PubMed PMID: 32072615]
[26]
Terrier B, Jaccard A, Harousseau JL, Delarue R, Tournilhac O, Hunault-Berger M, Hamidou M, Dantal J, Bernard M, Grosbois B, Morel P, Coiteux V, Gisserot O, Rodon P, Hot A, Elie C, Leblond V, Fermand JP, Fakhouri F. The clinical spectrum of IgM-related amyloidosis: a French nationwide retrospective study of 72 patients. Medicine. 2008 Mar:87(2):99-109. doi: 10.1097/MD.0b13e31816c43b6. Epub
[PubMed PMID: 18344807]
Level 2 (mid-level) evidence
[27]
Gertz MA. Immunoglobulin light chain amyloidosis: 2018 Update on diagnosis, prognosis, and treatment. American journal of hematology. 2018 Sep:93(9):1169-1180. doi: 10.1002/ajh.25149. Epub
[PubMed PMID: 30040145]
[28]
Desai HV, Aronow WS, Peterson SJ, Frishman WH. Cardiac amyloidosis: approaches to diagnosis and management. Cardiology in review. 2010 Jan-Feb:18(1):1-11. doi: 10.1097/CRD.0b013e3181bdba8f. Epub
[PubMed PMID: 20010333]
[29]
Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, Laumann K, Zeldenrust SR, Leung N, Dingli D, Greipp PR, Lust JA, Russell SJ, Kyle RA, Rajkumar SV, Gertz MA. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012 Mar 20:30(9):989-95. doi: 10.1200/JCO.2011.38.5724. Epub 2012 Feb 13
[PubMed PMID: 22331953]