Continuing Education Activity
Acrokeratoelastoidosis is a rare, benign, and generally asymptomatic condition that usually begins in childhood and is characterized by flesh-colored papules on the lateral areas of the palms, soles, and dorsum of hands. Treatment options have only shown marginal efficacy, however, practitioners should be familiar with this condition so that it can be evaluated properly and other, more concerning forms of ketatodermas that may suggest the possibility of some serious internal disorder or malignancy can be ruled out. This activity reviews the evaluation and management of patients with acrokeratoelastoidosis and highlights the role of the interprofessional team in caring for patients with this condition.
Objectives:
- Describe the clinical presentation, epidemiology, and probable etiopathogenesis of acrokeratoelastoidosis.
- Outline the diagnostic approach and treatment options for acrokeratoelastoidosis.
- Summarize the expected course and prognosis of acrokeratoelastoidosis and the education of the patient and family about the benign nature of the condition, how to alleviate their anxiety, and explain what to expect from treatment.
- Explain the importance of a cohesive, interprofessional team approach when counseling a patient and his or her family about the benign nature and good prognosis of the condition.
Introduction
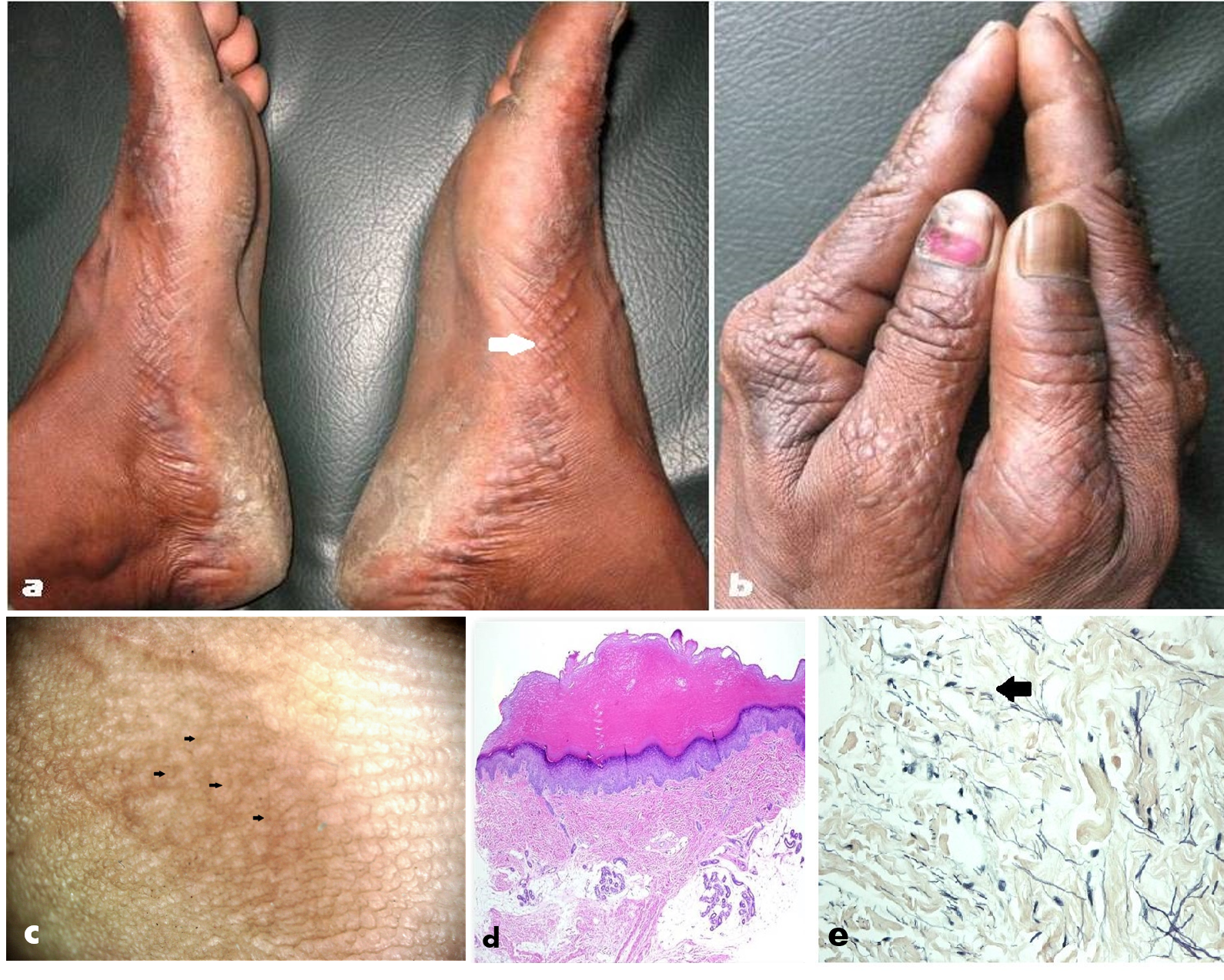
Acrokeratoelastoidosis (AKE) of Oswaldo Costa, or inverse papular acrokeratosis, is a rare autosomal-dominant genodermatosis first described in 1952 by Oswaldo Costa, a Brazilian dermatologist.[1] It characteristically presents with flesh-colored papules on the lateral areas of the palms, soles, and dorsum of hands. Acrokeratoelastoidosis is a type of marginal keratoderma that principally affects the lateral portion of the palmoplantar regions [Figures 1 (A) and (B)]. Apart from inherited cases, there are reports of sporadic occurrences of this condition.[2] The histological features are hyalinized and homogenous collagen, hyperkeratosis, and a decrease in, and fragmentation of the elastic fibers (elastorrhexis).[3] This rare form of focal acral keratoderma of unknown cause typically begins during childhood, although onset may delay until adolescence. As per the limited published literature on this condition, there seems to be no gender or racial predilection. Another reason for some patients seeking a dermatologist's opinion a few years after the onset is the asymptomatic nature of the lesions. The condition requires differentiation from two sets of disorders: 1) other marginal and focal acral keratodermas, and 2) distinct disorders such as acrokeratosis verruciformis of Hopf. Histopathological findings aid in differentiation between acrokeratoelastoidosis and other clinical simulators. There is no reported morbidity, and apart from cosmetic concerns of some patients, the overall prognosis of AKE is good.[3][4]
Etiology
Genetic Factors
Although there are reports of sporadic cases, acrokeratoelastoidosis is considered to be a genodermatoses with autosomal dominant being the primary mode of inheritance (although anecdotal reports exist of an autosomal recessive mode). As per the classification of palmoplantar keratodermas (PPK), inherited acrokeratoelastoidosis is considered to be a type of inherited punctate palmoplantar keratoderma (PPKP), specifically Type 3 PPKP.[5][6] OMIM recognizes three distinct types of inherited punctate PPK. These are; PPKP1 (also known as the Buschke-Fischer-Brauer type, OMIM #148600), mapped to 15q22; PPKP2 (porokeratotic type, OMIM #175860), not yet mapped; and PPKP3 (acrokeratoelastoidosis, OMIM #101850), where preliminary linkage suggests a possible locus on 2p25-p12.[5] Thus, chromosome 2 seems the most probable locus involved in inherited AKE.[7] It is important to mention that the AAGAB gene, implicated in type I PPK, is not involved in AKE.[6] In PPKP1, AAGAB gene associated reduced expression of the cytosolic p34 protein has been implicated, which results in increased epidermal cell proliferation.[5][8] The possibility of a similar gene-protein complex involved in the pathogenesis of acrokeratoelastoidosis cannot be discounted and merits further exploration.
Acquired Factors
The exact pathogenesis remains unclear. Although a history of chronic trauma and excessive sun-exposure has been reported in few cases[2][3][4][9][10]; there is no establishment of a direct causal relationship.
It is important to note that although the clinical presentation is predominated by lesions generally localized to the acral parts; some workers have reported that elastorrhexis (fragmentation of dermal elastic fibers). The histopathological hallmark of the condition seen in acral papules also involves apparently normal-appearing skin in some patients suggesting that AKE may represent a disorder with a generalized defect of elastic tissue.[11] Presumptively, factors like chronic or repeated bouts of unnoticed trauma (which are much more common over the acral parts of the body) may result in the predominance of clinical expression of the disorder over the palms and soles.
The anecdotal report of AKE-like lesions in patients with scleroderma has its basis in the abnormal connective tissue metabolism of scleroderma.[12][13] Other anecdotal observations of unclear etiological association include hyperhidrosis and aquagenic PPK.[10][14][15]
Epidemiology
The disease is not congenital; instead, its typical onset occurs in childhood or adolescence. As mentioned before, evaluation of the limited number of total reported cases does not suggest a predilection for any gender or ethnicity. Owing to the rarity of the condition, the exact incidence of the disorder remains unknown.
Most cases of AKEs that have been reported so far had their onset before the second and third decades of life. The youngest age reported for a patient of AKE was 3 years.[9]
Although bilateral involvement is characteristic of acrokeratoelastoidosis, there are reports of a few cases with unilateral involvemen.[2][16] There have been suggestions that the unilateral variant of AKE arises from genetic mosaicism.
Although some investigators have reported an increase in the number of papules over time,[10] a general consensus to that observation remains elusive.
Pathophysiology
In familial cases that constitute the majority of the reports to date implicate a genetic abnormality involving chromosome 2. A hypothesis of altered expression of a gene-protein complex akin to p34 involvement in PPKP1 has been previously mentioned (vide supra). In sporadic cases, factors like excessive sun-exposure and chronic trauma may serve as possible triggering factors.
Elastorrhexis is the histo-pathophysiological hallmark of the disorder. Exaggerated production and accumulation of filaggrin in the form of a dense band over stratum granulosum, before getting incorporated into the protein matrix of mature epidermal keratin has been postulated to result in the formation of the characteristic keratotic papules.[17] Presence of abnormal dense granules in dermal fibroblasts of lesional skin hints at the possibility of acrokeratoelastoidosis representing the outcome of abnormal secretion of elastic fibers rather than that of fiber degradation as its name suggests.[17]
Histopathology
The most common findings on histopathology [Figure 1 (D)] are hyperkeratosis, hypergranulosis, mild acanthosis, collagen homogenization and alterations in the elastic fibers of the dermis, which are fewer and fragmented (elastorrhexis).[3] Hyperkeratosis sometimes causes depression in the underlying epithelial planes, forming a concavity.[3] One observes areas of collagen homogenization in the superficial dermis with thin elastic and reduced and fragmented fibers.
It is noteworthy that elastorrhexis involving the dermis down to the reticular dermis level is a unique and archetypical pathological feature of acrokeratoelastoidosis, which also differentiates it from acral focal hyperkeratosis, a very close clinical differential. In the latter, histological alterations are limited to the epidermis in the form of epidermal hyperkeratosis with elastorrhexis of the reticular dermis typically absent.[18]
Electron microscopy shows disaggregation of elastic fibers with microfibrillar fragmentation.[18]
Special stains for elastin (and collagen) such as VVG, Weigert, and oOrcein demonstrate the changes in elastic fibers (elastorrhexis) better [Figure 1(E)] than the routine hematoxylin-eosin stain.[3]
History and Physical
Acrokeratoelastoidosis typically presents in children and adolescents, usually before the 2nd and 3rd decades of life, although reports do exist of onset in adulthood.[19] Clinically, AKE presents with clusters of multiple asymptomatic, small, round-to-oval, skin-colored/translucent or yellowish firm papules located over the lateral and/or medial margins of hands and/or feet. Papules have a keratotic rough surface and may show a crateriform to umbilicated appearance [Figure 1(A)]. Papular acrokeratosis, a synonym for acrokeratoelastoidosis stems from such characteristic morphology and location of lesions. Involvement of the dorsa of hands/feet is characteristic [Figure 1(B)], qualifying the lesion as 'inverse' papular acrokeratosis.[18][20] Other areas reported to have occasional involvement include - the posteromedial border of the feet and the pre-tibial region.[20] The lesions are typically distributed in a bilateral and symmetrical pattern, although unilateral involvement has been reported.[2][16][21] The papules may coalesce to form plaques.
Typically, except for cosmetic disfigurement, the lesions are asymptomatic in most of the cases. Mild itching, hyperhidrosis, and aquagenic palmoplantar keratoderma constitute uncommon presentations/associations.[3][15] A family history of similar lesions is commonly positive since the majority of cases show an autosomal dominant mode of inheritance. The clinician should inquire about a history of chronic or repeated trauma to palms and soles or excessive sun exposure. Trauma, handwashing, clothes, and irritation from ill-fitting footwear may increase the dimensions of acrokeratoelastoidosis.
In most cases, the lesions stabilize after a few weeks to months of onset although there are anecdotal reports of rapid progression of lesions during pregnancy.[10]
Evaluation
Dermoscopy
Polarized videodermoscopic evaluation of the affected areas of the thumbs and index fingers shows focal clusters of pale-to-yellowish colored papules with slight umbilication in a few of them, interspersed with pale yellow-colored structureless areas [Figure 1 (C)].
Histopathology
The gold-standard technique of diagnosis of acrokeratoelastoidosis remains histopathology. Apart from various epidermal alterations (vide infra), the presence of a reduced number of thick and fragmented elastic fibers in the dermis ('elastorrhexis') is the diagnostic hallmark; the latter feature is observed even on a routine hematoxylin-eosin (H & E) section [Figure 1 (D)].but better appreciated with special stains for elastin such as Verhoeff’s-Van Gieson (VVG) [Figure 1 (E)], Weigert and Orcein.
Ultrasonography: Doppler Study
In a recent study, the use of Doppler ultrasound (USG) examination was reported to be a useful non-invasive adjuvant investigation for confirmation of diagnosis and benignancy of the condition (lack of protrusions of fat lobules) and ruling out the presence of piezogenic pedal papules and granulomas.[22] In acrokeratoelastoidosis, doppler USG demonstrates several focal solid hypoechoic and hypovascular areas with thickening of the epidermis and upper dermis.[22]
Treatment / Management
Since the condition is asymptomatic and benign, treatment should focus on patients who are cosmetically concerned. To date, various forms of treatments - topical, oral and surgical/physical have been attempted to improve upon the appearance of AKE-affected acral parts of the patients, but the outcome has been modest to poor. The various treatment modalities tried [3][10][22][10][23][24]:
- Topical - emollients and keratolytic agents including salicylic acid, urea, sulfur, coal tar, and tretinoin, as well as topical corticosteroids
- Oral - corticosteroids, antibiotics, dapsone, methotrexate, isotretinoin, acitretin
- Physical/surgical - cryotherapy with liquid nitrogen, and surgery with laser erbium: yttrium-aluminum-garnet (Er: YAG)
Treatment with oral retinoids, especially acitretin has been suggested as the most effective; however, relapse post-cessation is inevitable[10]
Summarily, even in patients with acrokeratoelastoidosis offered treatment for cosmetic improvement, three factors merit consideration and require explanation to the patient before commencing treatment:
- Modest response to be expected
- If using topicals - indefinite use may be necessary to maintain any improvement
- If planning oral/systemic therapy - the possibility of an almost inevitable relapse on stoppage of the drug, and the adverse effects of the drug(s)
Differential Diagnosis
Differential diagnosis includes conditions considered as members of the family of marginal and acral keratodermas, and other distinctive disorders involving the acral parts with similar appearing lesions. It is important to know that involvement of the borders or palmoplantar transition areas typifies 'marginal keratodermas'; involvement of the dorsum of the hands and feet qualifies the marginal keratoderma to be called 'inverse.'[18] Some of the important differentials include[3][10][25][22]:
- Focal acral hyperkeratosis - differentiable on histopathology due to changes limited to the epidermis, with the absence of elastorrhexis
- Keratoelastoidosis marginalis (also known as Ramos and Silva marginal keratoderma) - associated with intense sun exposure and shows prominent actinic damage
- Other acral keratoderma variants - hereditary papulotranslucent acrokeratoderma, acrokeratoderma hereditarium punctatum, punctate palmoplantar keratoderma
- Miscellaneous conditions - acrokeratosis verruciformis of Hopf, degenerative collagenous plaques (of the hands), digital papular calcinosis, verruca plana, primary cutaneous amyloidosis, and mosaic acral keratosis
Prognosis
It is important to reiterate that many forms of PPK, including certain types of PPKP, are harbingers or cutaneous markers of serious underlying morbidity. However, AKE is a benign form of PPKP, with no associated medical condition. Discounting the fact of poor response to treatments and indefinite persistence of lesions, the overall prognosis of the condition is excellent.
Complications
There are no known or reported complications of acrokeratoelastoidosis.
Deterrence and Patient Education
Despite the good prognosis of this condition, many issues trouble the patient and family members, such as the cosmetic disfigurement caused by the papules, persistence of lesions and modest-to-poor response of treatments tried until date for acrokeratoelastoidosis of Costa. Thus, patient and family education about the benignity of the condition constitutes a much more critical component of management than suggesting treatment for cosmetic improvement of the lesions (vide supra).
Although AKE is a simple clinical diagnosis, it may be logical to get a skin biopsy done followed by re-affirmation of the condition's benign nature. The general perception in the mind of patients of 'biopsy is suggestive of an ominous disorder like a malignancy' is convincingly contradicted with the biopsy report of AKE revealing benign features. Of course, the treating dermatologist's counseling skills constitute an essential part of adequate patient education.
Pearls and Other Issues
Acrokeratoelastoidosis of Costa is a type of marginal keratoderma that typically affects the borders of palms and soles and frequently involves the dorsum of hands and feet.
In most cases, it is of familial origin with an autosomal-dominant mode of inheritance (although rarely it may be autosomal recessive). However sporadic occurrence is well known.
Clusters of skin-colored to yellowish-brown keratotic papules appear along the borders of hands and feet, typically during the 2nd and 3rd decade of life. The papules often have an umbilicated or crateriform appearance.
Chronic trauma and excessive sun exposure have been proposed as possible exacerbating factors.
Despite the condition's rarity, detections is straightforward based on clinical features alone. However, diagnostic confirmation warrants histopathology. Elastorrhexis, i.e., fragmented elastic fibers in the dermis constitutes the pathological diagnostic hallmark, better appreciated on slides specially stained to highlight elastin such as VVG or Orcein.
Treatment is required only for cosmetic reasons. Attempts with topical corticosteroids, salicylic acid, urea, coal tar, oral acitretin, and surgical techniques including liquid nitrogen cryotherapy, and erbium: yttrium-aluminum-garnet (Er: YAG) laser ablation have all shown only a modest and temporary improvement.
Oral retinoids provide the best improvement, but relapse following cessation of therapy and the potential adverse effects of the systemic therapy for an otherwise benign condition do not support their liberal use.
The most important part of management is patient and family counseling about the benign nature and good prognosis of the condition.
Enhancing Healthcare Team Outcomes
Owing to the rarity of this genodermatoses, two issues are potentially problematic. Firstly, non-dermatologists' awareness about this condition is very low and needs to be enhanced. This CME activity is a humble attempt towards that. Secondly, the treatment-related evidence for acrokeratoelastoidosis is limited to anecdotal reports only. Since controlled trials seem impractical because of the condition's rarity, small-to-large case series with a single intervention should be the way forward.