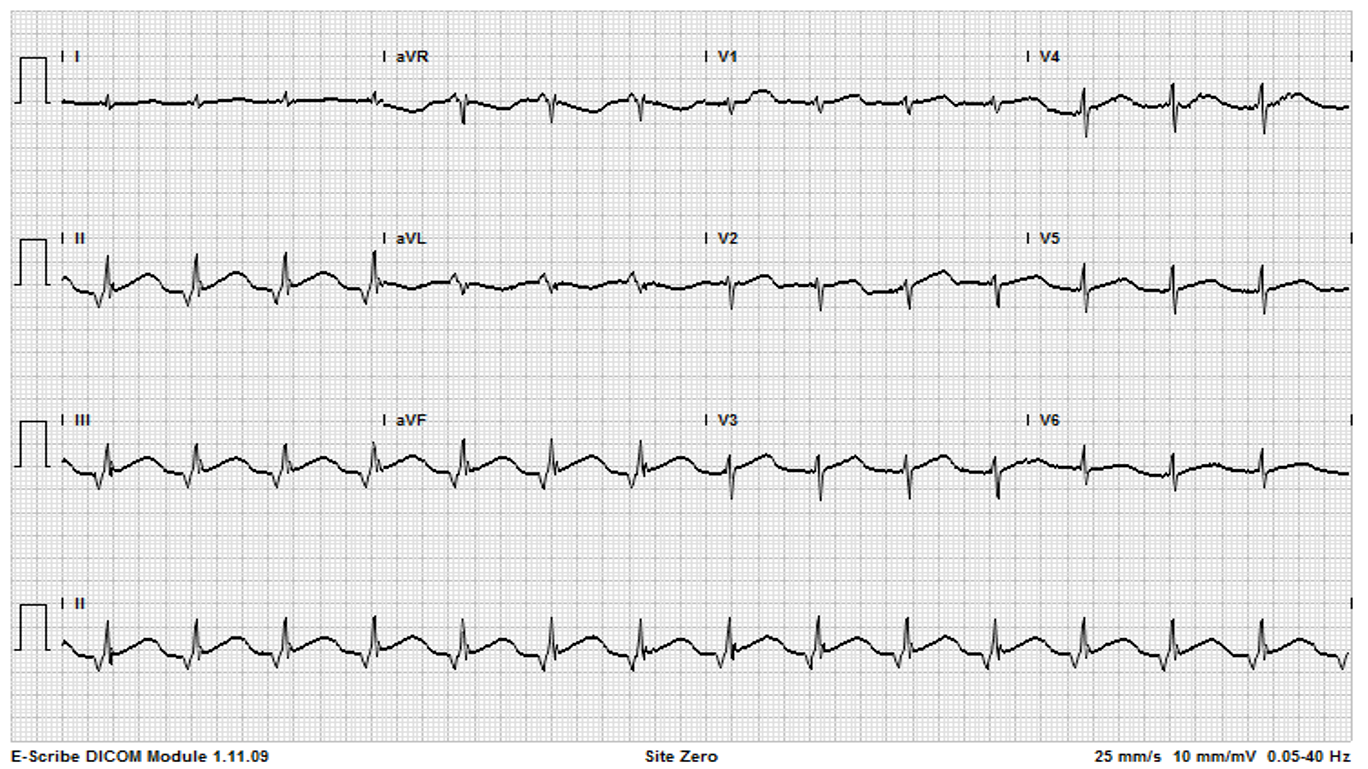
[1]
Zhou X, Bueno-Orovio A, Schilling RJ, Kirkby C, Denning C, Rajamohan D, Burrage K, Tinker A, Rodriguez B, Harmer SC. Investigating the Complex Arrhythmic Phenotype Caused by the Gain-of-Function Mutation KCNQ1-G229D. Frontiers in physiology. 2019:10():259. doi: 10.3389/fphys.2019.00259. Epub 2019 Mar 18
[PubMed PMID: 30967788]
[2]
Dehghani-Samani A, Madreseh-Ghahfarokhi S, Dehghani-Samani A. Mutations of Voltage-Gated Ionic Channels and Risk of Severe Cardiac Arrhythmias. Acta Cardiologica Sinica. 2019 Mar:35(2):99-110. doi: 10.6515/ACS.201903_35(2).20181028A. Epub
[PubMed PMID: 30930557]
[3]
Alahmadi A, Davies A, Vigo M, Jay C. Can laypeople identify a drug-induced QT interval prolongation? A psychophysical and eye-tracking experiment examining the ability of nonexperts to interpret an ECG. Journal of the American Medical Informatics Association : JAMIA. 2019 May 1:26(5):404-411. doi: 10.1093/jamia/ocy183. Epub
[PubMed PMID: 30848818]
[4]
Giudicessi JR,Roden DM,Wilde AAM,Ackerman MJ, Classification and Reporting of Potentially Proarrhythmic Common Genetic Variation in Long QT Syndrome Genetic Testing. Circulation. 2018 Feb 6;
[PubMed PMID: 29431662]
[5]
Beach SR, Celano CM, Sugrue AM, Adams C, Ackerman MJ, Noseworthy PA, Huffman JC. QT Prolongation, Torsades de Pointes, and Psychotropic Medications: A 5-Year Update. Psychosomatics. 2018 Mar-Apr:59(2):105-122. doi: 10.1016/j.psym.2017.10.009. Epub 2017 Nov 3
[PubMed PMID: 29275963]
[7]
Ebrahim MA, Williams MR, Shepard S, Perry JC. Genotype Positive Long QT Syndrome in Patients With Coexisting Congenital Heart Disease. The American journal of cardiology. 2017 Jul 15:120(2):256-261. doi: 10.1016/j.amjcard.2017.04.018. Epub 2017 Apr 27
[PubMed PMID: 28532774]
[8]
Uvelin A,Pejaković J,Mijatović V, Acquired prolongation of QT interval as a risk factor for torsade de pointes ventricular tachycardia: a narrative review for the anesthesiologist and intensivist. Journal of anesthesia. 2017 Jun;
[PubMed PMID: 28229241]
Level 3 (low-level) evidence
[9]
Hutton DM. The importance of routine QT interval measurement in rhythm interpretation. Dynamics (Pembroke, Ont.). 2008 Fall:19(3):29-33
[PubMed PMID: 18773713]
[10]
Cavero I, Crumb W. The use of electrocardiograms in clinical trials: a public discussion of the proposed ICH E14 regulatory guidance. April 11-12, 2005, Bethesda, MD, USA. Expert opinion on drug safety. 2005 Jul:4(4):795-9
[PubMed PMID: 16011455]
Level 3 (low-level) evidence
[11]
Anderson HN, Bos JM, Haugaa KH, Morlan BW, Tarrell RF, Caraballo PJ, Ackerman MJ. Prevalence and Outcome of High-Risk QT Prolongation Recorded in the Emergency Department from an Institution-Wide QT Alert System. The Journal of emergency medicine. 2018 Jan:54(1):8-15. doi: 10.1016/j.jemermed.2017.08.073. Epub 2017 Oct 26
[PubMed PMID: 29107482]
[12]
Gromova OA,Torshin IY,Kalacheva AG,Grishina TR, [On Synergism of Potassium and Magnesium in Maintenance of Myocardial Function]. Kardiologiia. 2016 Mar;
[PubMed PMID: 28294893]
[13]
Chouchoulis K, Chiladakis J, Koutsogiannis N, Davlouros P, Kaza M, Alexopoulos D. Impact of QT interval prolongation following antiarrhythmic drug therapy on left ventricular function. Future cardiology. 2017 Jan:13(1):13-22. doi: 10.2217/fca-2016-0052. Epub 2016 Dec 19
[PubMed PMID: 27990843]