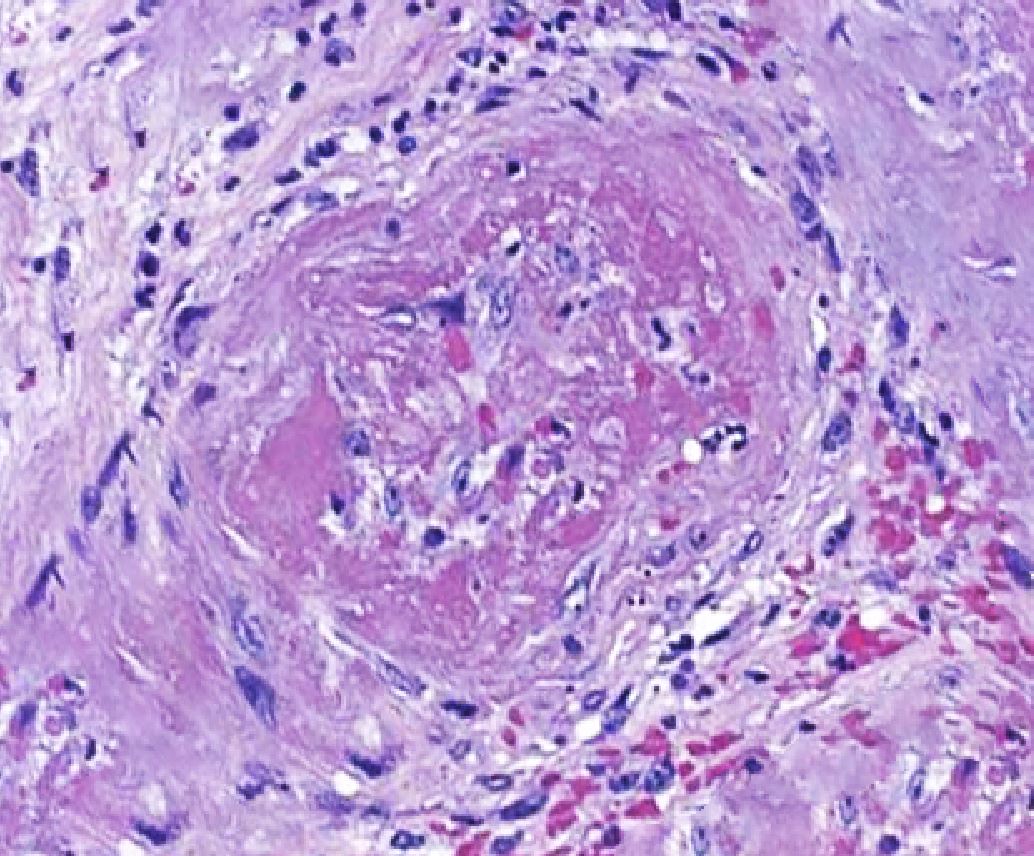
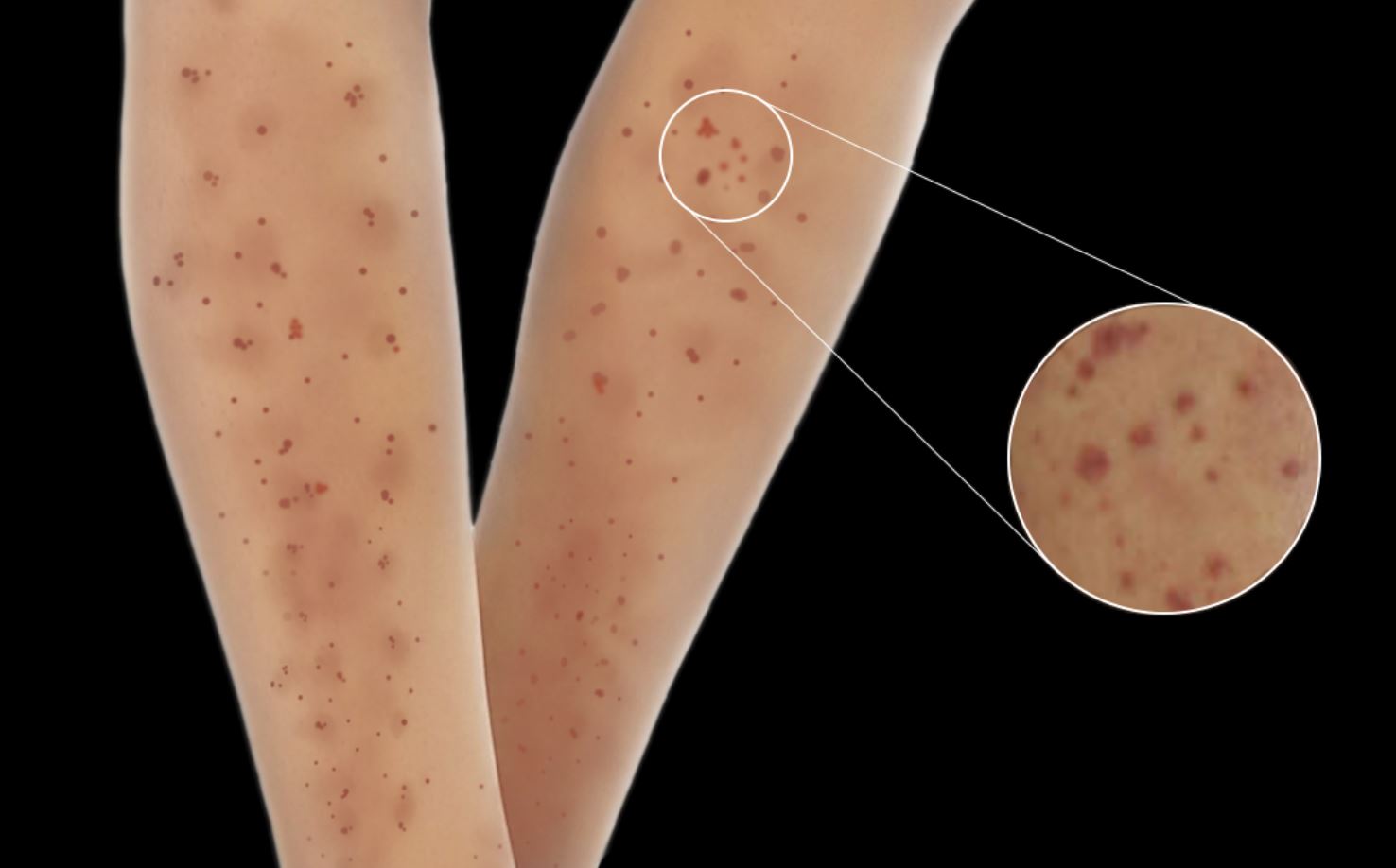
[1]
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores-Suarez LF, Gross WL, Guillevin L, Hagen EC, Hoffman GS, Jayne DR, Kallenberg CG, Lamprecht P, Langford CA, Luqmani RA, Mahr AD, Matteson EL, Merkel PA, Ozen S, Pusey CD, Rasmussen N, Rees AJ, Scott DG, Specks U, Stone JH, Takahashi K, Watts RA. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis and rheumatism. 2013 Jan:65(1):1-11. doi: 10.1002/art.37715. Epub
[PubMed PMID: 23045170]
Level 3 (low-level) evidence
[2]
Chung SA, Seo P. Microscopic polyangiitis. Rheumatic diseases clinics of North America. 2010 Aug:36(3):545-58. doi: 10.1016/j.rdc.2010.04.003. Epub 2010 Jun 11
[PubMed PMID: 20688249]
[3]
Greco A, Rizzo MI, De Virgilio A, Gallo A, Fusconi M, Pagliuca G, Martellucci S, Turchetta R, Longo L, De Vincentiis M. Goodpasture's syndrome: a clinical update. Autoimmunity reviews. 2015 Mar:14(3):246-53. doi: 10.1016/j.autrev.2014.11.006. Epub 2014 Nov 15
[PubMed PMID: 25462583]
[4]
Kallenberg CG,Heeringa P,Stegeman CA, Mechanisms of Disease: pathogenesis and treatment of ANCA-associated vasculitides. Nature clinical practice. Rheumatology. 2006 Dec
[PubMed PMID: 17133251]
[5]
de Lind van Wijngaarden RA, van Rijn L, Hagen EC, Watts RA, Gregorini G, Tervaert JW, Mahr AD, Niles JL, de Heer E, Bruijn JA, Bajema IM. Hypotheses on the etiology of antineutrophil cytoplasmic autoantibody associated vasculitis: the cause is hidden, but the result is known. Clinical journal of the American Society of Nephrology : CJASN. 2008 Jan:3(1):237-52
[PubMed PMID: 18077783]
[6]
Pendergraft WF 3rd, Niles JL. Trojan horses: drug culprits associated with antineutrophil cytoplasmic autoantibody (ANCA) vasculitis. Current opinion in rheumatology. 2014 Jan:26(1):42-9. doi: 10.1097/BOR.0000000000000014. Epub
[PubMed PMID: 24276086]
Level 3 (low-level) evidence
[7]
Wakamatsu K, Mitsuhashi Y, Yamamoto T, Tsuboi R. Propylthiouracil-induced antineutrophil cytoplasmic antibody positive vasculitis clinically mimicking pyoderma gangrenosum. The Journal of dermatology. 2012 Aug:39(8):736-8. doi: 10.1111/j.1346-8138.2011.01399.x. Epub 2011 Dec 2
[PubMed PMID: 22132781]
[8]
Lyons PA,Rayner TF,Trivedi S,Holle JU,Watts RA,Jayne DR,Baslund B,Brenchley P,Bruchfeld A,Chaudhry AN,Cohen Tervaert JW,Deloukas P,Feighery C,Gross WL,Guillevin L,Gunnarsson I,Harper L,Hrušková Z,Little MA,Martorana D,Neumann T,Ohlsson S,Padmanabhan S,Pusey CD,Salama AD,Sanders JS,Savage CO,Segelmark M,Stegeman CA,Tesař V,Vaglio A,Wieczorek S,Wilde B,Zwerina J,Rees AJ,Clayton DG,Smith KG, Genetically distinct subsets within ANCA-associated vasculitis. The New England journal of medicine. 2012 Jul 19
[PubMed PMID: 22808956]
[9]
Vega Miranda J, Pinto Peñaranda LF, Márquez Hernández JD, Velásquez Franco CJ. Microscopic polyangiitis secondary to silica exposure. Reumatologia clinica. 2014 May-Jun:10(3):180-2. doi: 10.1016/j.reuma.2013.04.009. Epub 2013 Jul 23
[PubMed PMID: 23886979]
[10]
Berti A, Cornec D, Crowson CS, Specks U, Matteson EL. The Epidemiology of Antineutrophil Cytoplasmic Autoantibody-Associated Vasculitis in Olmsted County, Minnesota: A Twenty-Year US Population-Based Study. Arthritis & rheumatology (Hoboken, N.J.). 2017 Dec:69(12):2338-2350. doi: 10.1002/art.40313. Epub 2017 Nov 9
[PubMed PMID: 28881446]
[11]
Lane SE, Watts R, Scott DG. Epidemiology of systemic vasculitis. Current rheumatology reports. 2005 Aug:7(4):270-5
[PubMed PMID: 16045829]
[12]
Gonzalez-Gay MA, Garcia-Porrua C, Guerrero J, Rodriguez-Ledo P, Llorca J. The epidemiology of the primary systemic vasculitides in northwest Spain: implications of the Chapel Hill Consensus Conference definitions. Arthritis and rheumatism. 2003 Jun 15:49(3):388-93
[PubMed PMID: 12794795]
Level 3 (low-level) evidence
[13]
Mohammad AJ, Jacobsson LT, Mahr AD, Sturfelt G, Segelmark M. Prevalence of Wegener's granulomatosis, microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss syndrome within a defined population in southern Sweden. Rheumatology (Oxford, England). 2007 Aug:46(8):1329-37
[PubMed PMID: 17553910]
[14]
Flossmann O, Berden A, de Groot K, Hagen C, Harper L, Heijl C, Höglund P, Jayne D, Luqmani R, Mahr A, Mukhtyar C, Pusey C, Rasmussen N, Stegeman C, Walsh M, Westman K, European Vasculitis Study Group. Long-term patient survival in ANCA-associated vasculitis. Annals of the rheumatic diseases. 2011 Mar:70(3):488-94. doi: 10.1136/ard.2010.137778. Epub 2010 Nov 24
[PubMed PMID: 21109517]
[15]
Hagen EC, Daha MR, Hermans J, Andrassy K, Csernok E, Gaskin G, Lesavre P, Lüdemann J, Rasmussen N, Sinico RA, Wiik A, van der Woude FJ. Diagnostic value of standardized assays for anti-neutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. EC/BCR Project for ANCA Assay Standardization. Kidney international. 1998 Mar:53(3):743-53
[PubMed PMID: 9507222]
[16]
Kallenberg CG,Stegeman CA,Abdulahad WH,Heeringa P, Pathogenesis of ANCA-associated vasculitis: new possibilities for intervention. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2013 Dec
[PubMed PMID: 23810690]
[17]
Massicotte-Azarniouch D, Herrera CA, Jennette JC, Falk RJ, Free ME. Mechanisms of vascular damage in ANCA vasculitis. Seminars in immunopathology. 2022 May:44(3):325-345. doi: 10.1007/s00281-022-00920-0. Epub 2022 Mar 7
[PubMed PMID: 35254509]
[18]
Frankel SK, Schwarz MI. The pulmonary vasculitides. American journal of respiratory and critical care medicine. 2012 Aug 1:186(3):216-24. doi: 10.1164/rccm.201203-0539CI. Epub 2012 Jun 7
[PubMed PMID: 22679011]
[19]
Sun L, Wang H, Jiang X, Mo Y, Yue Z, Huang L, Liu T. Clinical and pathological features of microscopic polyangiitis in 20 children. The Journal of rheumatology. 2014 Aug:41(8):1712-9. doi: 10.3899/jrheum.131300. Epub 2014 Jul 1
[PubMed PMID: 24986845]
[20]
Hauer HA,Bajema IM,van Houwelingen HC,Ferrario F,Noël LH,Waldherr R,Jayne DR,Rasmussen N,Bruijn JA,Hagen EC, Renal histology in ANCA-associated vasculitis: differences between diagnostic and serologic subgroups. Kidney international. 2002 Jan
[PubMed PMID: 11786087]
[21]
Lababidi MH, Odigwe C, Okolo C, Elhassan A, Iroegbu N. Microscopic polyangiitis causing diffuse alveolar hemorrhage and rapidly progressive glomerulonephritis. Proceedings (Baylor University. Medical Center). 2015 Oct:28(4):469-71
[PubMed PMID: 26424944]
[22]
Wang H, Sun L, Tan W. Clinical features of children with pulmonary microscopic polyangiitis: report of 9 cases. PloS one. 2015:10(4):e0124352. doi: 10.1371/journal.pone.0124352. Epub 2015 Apr 29
[PubMed PMID: 25923706]
Level 3 (low-level) evidence
[23]
Wilke L, Prince-Fiocco M, Fiocco GP. Microscopic polyangiitis: a large single-center series. Journal of clinical rheumatology : practical reports on rheumatic & musculoskeletal diseases. 2014 Jun:20(4):179-82. doi: 10.1097/RHU.0000000000000108. Epub
[PubMed PMID: 24847742]
[24]
Karras A. Microscopic Polyangiitis: New Insights into Pathogenesis, Clinical Features and Therapy. Seminars in respiratory and critical care medicine. 2018 Aug:39(4):459-464. doi: 10.1055/s-0038-1673387. Epub 2018 Nov 7
[PubMed PMID: 30404112]
[25]
Barowka SE, Perrine TR, Hughes K. "I'm coughing up blood". Journal of the Mississippi State Medical Association. 2016 Nov:57(11):354-356
[PubMed PMID: 30281235]
[26]
Kluger N, Pagnoux C, Guillevin L, Francès C, French Vasculitis Study Group. Comparison of cutaneous manifestations in systemic polyarteritis nodosa and microscopic polyangiitis. The British journal of dermatology. 2008 Sep:159(3):615-20. doi: 10.1111/j.1365-2133.2008.08725.x. Epub 2008 Jul 19
[PubMed PMID: 18647311]
[27]
Lhote F, Cohen P, Guillevin L. Polyarteritis nodosa, microscopic polyangiitis and Churg-Strauss syndrome. Lupus. 1998:7(4):238-58
[PubMed PMID: 9643314]
[28]
Gibson LE, Cutaneous manifestations of antineutrophil cytoplasmic antibody-associated vasculitis (AAV): a concise review with emphasis on clinical and histopathologic correlation. International journal of dermatology. 2022 May 22
[PubMed PMID: 35599359]
[29]
Eriksson P, Segelmark M, Hallböök O. Frequency, Diagnosis, Treatment, and Outcome of Gastrointestinal Disease in Granulomatosis with Polyangiitis and Microscopic Polyangiitis. The Journal of rheumatology. 2018 Apr:45(4):529-537. doi: 10.3899/jrheum.170249. Epub 2018 Feb 1
[PubMed PMID: 29419474]
[30]
Hattori N, Mori K, Misu K, Koike H, Ichimura M, Sobue G. Mortality and morbidity in peripheral neuropathy associated Churg-Strauss syndrome and microscopic polyangiitis. The Journal of rheumatology. 2002 Jul:29(7):1408-14
[PubMed PMID: 12136898]
[31]
Xu J, Ding Y, Qu Z, Yu F. Posterior Reversible Encephalopathy Syndrome in a Patient With Microscopic Polyangiitis: A Case Report and Literature Review. Frontiers in medicine. 2021:8():792744. doi: 10.3389/fmed.2021.792744. Epub 2021 Dec 24
[PubMed PMID: 35071272]
Level 3 (low-level) evidence
[32]
Ku BD,Shin HY, Multiple bilateral non-hemorrhagic cerebral infarctions associated with microscopic polyangiitis. Clinical neurology and neurosurgery. 2009 Dec
[PubMed PMID: 19733002]
[33]
Eschun GM, Mink SN, Sharma S. Pulmonary interstitial fibrosis as a presenting manifestation in perinuclear antineutrophilic cytoplasmic antibody microscopic polyangiitis. Chest. 2003 Jan:123(1):297-301
[PubMed PMID: 12527637]
[34]
Iida T, Adachi T, Tabeya T, Nakagaki S, Yabana T, Goto A, Kondo Y, Kasai K. Rare type of pancreatitis as the first presentation of anti-neutrophil cytoplasmic antibody-related vasculitis. World journal of gastroenterology. 2016 Feb 21:22(7):2383-90. doi: 10.3748/wjg.v22.i7.2383. Epub
[PubMed PMID: 26900301]
[35]
Sassi SB, Ghorbel IB, Mizouni H, Houman MH, Hentati F. Microscopic polyangiitis presenting with peripheral and central neurological manifestations. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2011 Aug:32(4):727-9. doi: 10.1007/s10072-011-0653-x. Epub 2011 Jun 17
[PubMed PMID: 21681367]
[36]
Nachman PH,Hogan SL,Jennette JC,Falk RJ, Treatment response and relapse in antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. Journal of the American Society of Nephrology : JASN. 1996 Jan
[PubMed PMID: 8808107]
[37]
Crickx E, Machelart I, Lazaro E, Kahn JE, Cohen-Aubart F, Martin T, Mania A, Hatron PY, Hayem G, Blanchard-Delaunay C, de Moreuil C, Le Guenno G, Vandergheynst F, Maurier F, Crestani B, Dhote R, Silva NM, Ollivier Y, Mehdaoui A, Godeau B, Mariette X, Cadranel J, Cohen P, Puéchal X, Le Jeunne C, Mouthon L, Guillevin L, Terrier B, French Vasculitis Study Group. Intravenous Immunoglobulin as an Immunomodulating Agent in Antineutrophil Cytoplasmic Antibody-Associated Vasculitides: A French Nationwide Study of Ninety-Two Patients. Arthritis & rheumatology (Hoboken, N.J.). 2016 Mar:68(3):702-12. doi: 10.1002/art.39472. Epub
[PubMed PMID: 26473632]
[38]
de Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL, Luqmani R, Pusey CD, Rasmussen N, Sinico RA, Tesar V, Vanhille P, Westman K, Savage CO, EUVAS (European Vasculitis Study Group). Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Annals of internal medicine. 2009 May 19:150(10):670-80
[PubMed PMID: 19451574]
Level 1 (high-level) evidence
[39]
Frankel SK, Cosgrove GP, Fischer A, Meehan RT, Brown KK. Update in the diagnosis and management of pulmonary vasculitis. Chest. 2006 Feb:129(2):452-465. doi: 10.1378/chest.129.2.452. Epub
[PubMed PMID: 16478866]
[40]
Falk RJ, Jennette JC. ANCA disease: where is this field heading? Journal of the American Society of Nephrology : JASN. 2010 May:21(5):745-52. doi: 10.1681/ASN.2009121238. Epub 2010 Apr 15
[PubMed PMID: 20395376]
[41]
Smith RM, Jones RB, Jayne DR. Progress in treatment of ANCA-associated vasculitis. Arthritis research & therapy. 2012 Apr 30:14(2):210. doi: 10.1186/ar3797. Epub 2012 Apr 30
[PubMed PMID: 22569190]
[42]
Millet A, Pederzoli-Ribeil M, Guillevin L, Witko-Sarsat V, Mouthon L. Antineutrophil cytoplasmic antibody-associated vasculitides: is it time to split up the group? Annals of the rheumatic diseases. 2013 Aug:72(8):1273-9. doi: 10.1136/annrheumdis-2013-203255. Epub 2013 Apr 20
[PubMed PMID: 23606701]
[43]
Thurner L, Preuss KD, Fadle N, Regitz E, Klemm P, Zaks M, Kemele M, Hasenfus A, Csernok E, Gross WL, Pasquali JL, Martin T, Bohle RM, Pfreundschuh M. Progranulin antibodies in autoimmune diseases. Journal of autoimmunity. 2013 May:42():29-38. doi: 10.1016/j.jaut.2012.10.003. Epub 2012 Nov 11
[PubMed PMID: 23149338]
[44]
Specks U, Merkel PA, Seo P, Spiera R, Langford CA, Hoffman GS, Kallenberg CG, St Clair EW, Fessler BJ, Ding L, Viviano L, Tchao NK, Phippard DJ, Asare AL, Lim N, Ikle D, Jepson B, Brunetta P, Allen NB, Fervenza FC, Geetha D, Keogh K, Kissin EY, Monach PA, Peikert T, Stegeman C, Ytterberg SR, Mueller M, Sejismundo LP, Mieras K, Stone JH, RAVE-ITN Research Group. Efficacy of remission-induction regimens for ANCA-associated vasculitis. The New England journal of medicine. 2013 Aug 1:369(5):417-27. doi: 10.1056/NEJMoa1213277. Epub
[PubMed PMID: 23902481]
[45]
Jayne D, Rasmussen N, Andrassy K, Bacon P, Tervaert JW, Dadoniené J, Ekstrand A, Gaskin G, Gregorini G, de Groot K, Gross W, Hagen EC, Mirapeix E, Pettersson E, Siegert C, Sinico A, Tesar V, Westman K, Pusey C, European Vasculitis Study Group. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. The New England journal of medicine. 2003 Jul 3:349(1):36-44
[PubMed PMID: 12840090]
Level 1 (high-level) evidence
[46]
Ward ND, Cosner DE, Lamb CA, Li W, Macknis JK, Rooney MT, Zhang PL. Top Differential Diagnosis Should Be Microscopic Polyangiitis in ANCA-Positive Patient with Diffuse Pulmonary Hemorrhage and Hemosiderosis. Case reports in pathology. 2014:2014():286030. doi: 10.1155/2014/286030. Epub 2014 Nov 30
[PubMed PMID: 25525543]
Level 3 (low-level) evidence
[47]
Chotiyarnwong P, McCloskey EV. Pathogenesis of glucocorticoid-induced osteoporosis and options for treatment. Nature reviews. Endocrinology. 2020 Aug:16(8):437-447. doi: 10.1038/s41574-020-0341-0. Epub 2020 Apr 14
[PubMed PMID: 32286516]
[48]
Haldar S, Dru C, Bhowmick NA. Mechanisms of hemorrhagic cystitis. American journal of clinical and experimental urology. 2014:2(3):199-208
[PubMed PMID: 25374922]
[49]
Kitching AR, Anders HJ, Basu N, Brouwer E, Gordon J, Jayne DR, Kullman J, Lyons PA, Merkel PA, Savage COS, Specks U, Kain R. ANCA-associated vasculitis. Nature reviews. Disease primers. 2020 Aug 27:6(1):71. doi: 10.1038/s41572-020-0204-y. Epub 2020 Aug 27
[PubMed PMID: 32855422]
[50]
Robson J, Doll H, Suppiah R, Flossmann O, Harper L, Höglund P, Jayne D, Mahr A, Westman K, Luqmani R. Glucocorticoid treatment and damage in the anti-neutrophil cytoplasm antibody-associated vasculitides: long-term data from the European Vasculitis Study Group trials. Rheumatology (Oxford, England). 2015 Mar:54(3):471-81. doi: 10.1093/rheumatology/keu366. Epub 2014 Sep 8
[PubMed PMID: 25205825]
[51]
Hassan TM, Hassan AS, Igoe A, Logan M, Gunaratnam C, McElvaney NG, O'Neill SJ. Lung involvement at presentation predicts disease activity and permanent organ damage at 6, 12 and 24 months follow - up in ANCA - associated vasculitis. BMC immunology. 2014 May 27:15():20. doi: 10.1186/1471-2172-15-20. Epub 2014 May 27
[PubMed PMID: 24884372]
[52]
Robson J, Doll H, Suppiah R, Flossmann O, Harper L, Höglund P, Jayne D, Mahr A, Westman K, Luqmani R. Damage in the anca-associated vasculitides: long-term data from the European vasculitis study group (EUVAS) therapeutic trials. Annals of the rheumatic diseases. 2015 Jan:74(1):177-84. doi: 10.1136/annrheumdis-2013-203927. Epub 2013 Nov 15
[PubMed PMID: 24243925]
[53]
Stone JH, Hoffman GS, Merkel PA, Min YI, Uhlfelder ML, Hellmann DB, Specks U, Allen NB, Davis JC, Spiera RF, Calabrese LH, Wigley FM, Maiden N, Valente RM, Niles JL, Fye KH, McCune JW, St Clair EW, Luqmani RA, International Network for the Study of the Systemic Vasculitides (INSSYS). A disease-specific activity index for Wegener's granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis and rheumatism. 2001 Apr:44(4):912-20
[PubMed PMID: 11318006]