Continuing Education Activity
Glycogen storage diseases (GSDs) are inherited inborn errors of carbohydrate metabolism. Clinical onset can range from neonatal life to adulthood. In general, they occur due to a lack of specific enzymes involved in the breakdown of glycogen and result in an abnormal buildup of glycogen in the liver or skeletal muscles. The inability to mobilize glucose from glycogen results in hypoglycemia and exercise-induced weakness in patients and leads to long-term complications. This activity describes the evaluation and management of GSDs and explains the interprofessional team's role in managing these patients.
Objectives:
- Identify the etiology of various glycogen storage diseases.
- Explain the typical patient history associated with glycogen storage diseases.
- Outline the diagnostic evaluation of patients suspected to have a glycogen storage disease.
- Summarize the importance of improving care coordination among the interprofessional team members to enhance the delivery of care for those with glycogen storage disease.
Introduction
Glycogen storage diseases (GSDs) are inherited inborn errors of carbohydrate metabolism. Disorders of carbohydrate metabolism that result in abnormal storage of glycogen are classified as GSDs. They are classified numerically in the order of recognition and identification of the enzyme defect causing the disorder. Clinical onset can range from neonatal life to adulthood. Depending on the specific type, GSDs can result from a failure to convert glycogen into energy and/or a toxic glycogen accumulation; however, all result in a failure to use or store glycogen.[1]
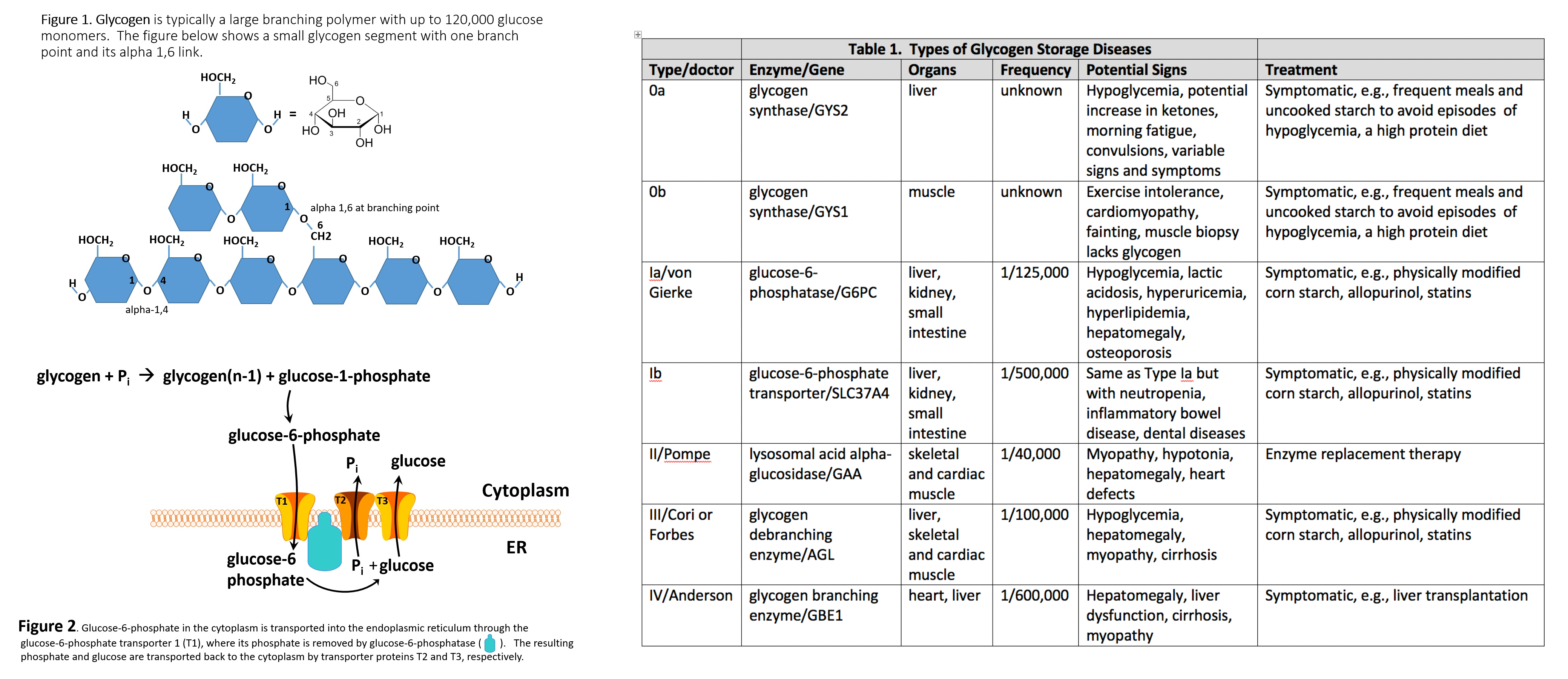
Glycogen is a branched polymer whose monomeric units are glucose (Figure 1). After a meal, the glucose level in plasma increases and stimulates the storage of excess glucose in cytoplasmic glycogen. The liver contains the highest percent of glycogen by weight (about 10%), whereas muscles can store about 2% by weight. Nevertheless, since the total muscle mass is greater than liver mass, the total mass of glycogen in the muscle is about twice that of the liver. When needed, the glycogen polymer can be broken down into glucose monomers and utilized for energy production. Many of the enzymes and transporters for these processes are key to the etiology of GSDs. An increasing number of GSDs are being identified, but most are very rare. Traditionally the GSDs were named after the clinician who first identified the disorder; however, they each have an identified enzyme and gene focus that will be used to refer to these disorders in this article, although the various diseases have their own classifications.[2][3]
Classification of Glycogen Storage Disorder
Glycogen storage disorders that primarily affect the liver include:
- Glycogen synthase-2 deficiency (GSD type 0a)
- Glucose-6-phosphatase deficiency (GSD type Ia)
- Glucose-6-phosphate transporter deficiency (GSD type Ib)
- Glycogen debrancher deficiency (GSD type III)
- Glycogen branching enzyme deficiency (GSD type IV)
- Liver phosphorylase deficiency (GSD type VI)
- Phosphorylase kinase deficiency (GSD type IXa)
- GLUT2 deficiency or Fanconi-Bickel disease
Glycogen storage disorders that primarily affect the skeletal muscles include:
- Muscle phosphorylase deficiency (GSD type V)
- Phosphofructokinase deficiency (GSD type VII)
- Phosphoglycerate mutase deficiency (GSD type X)
- Lactate dehydrogenase A deficiency (GSD type XI)
- Aldolase A deficiency (GSD type XII)
- Beta-enolase deficiency (GSD type XIII)
- Phosphoglucomutase-1 deficiency (GSD type XIV)
Glycogen storage disorders that affect both skeletal and cardiac muscles include:
- Lysosomal acid maltase deficiency (GSD type IIa)
- Lysosome-associated membrane protein 2 deficiency (GSD type IIb)
- Glycogenin-1 deficiency (GSD type XV)
- Muscle glycogen synthase deficiency (GSD type 0b)
Etiology
The etiology of GSDs is best understood by following the metabolic events leading to the synthesis (glycogenesis) and degradation of glycogen (glycogenolysis).[4] Excess dietary glucose is stored in glycogen, and glycogen synthesis is, in part, accomplished by glycogen synthase (GS). As indicated in Table 1, there are two distinct forms of glycogen synthase, one in the liver encoded by the GYS2 gene and one in skeletal muscle encoded by the GYS1 gene. Both forms of GS work by linking (alpha-1,4 links) a glucose monomer to the growing glycogen polymer. As indicated in Figure 1, glycogen has two different types of linkages, alpha-1,4 links and alpha-1,6 links.[4]
About 95% of linkages in glycogen are alpha-1,4 links.[4] The absence or malfunction of liver glycogen synthase due to mutations in the GYS2 gene will prevent glycogen from being synthesized in the liver. This is the cause of GSD type 0a. Similarly, the absence or malfunction of muscle glycogen synthase due to mutations in the GYS1 gene will prevent glycogen from being synthesized in muscles, and this is the cause of GSD type 0b.[2][3]
While glycogen synthase can catalyze the alpha-1,4 glucose linkages in glycogen, a different enzyme, glycogen branching enzyme (GBE1), is needed to produce the branching alpha-1,6 linkages. Mutations in the glycogen branching enzyme can result in the production of glycogen with an abnormal structure. This is the cause of GSD type IV.[2] The abnormal glycogen structures are called polyglucosan bodies. In GSD type IV, polyglucosan bodies accumulate in liver and muscle cells. Polyglucosan bodies do not effectively undergo glycogenolysis, and in muscle tissue, this can cause weakness and myopathy. In the liver, the accumulation of polyglucosan bodies causes hepatomegaly.[2]
While GSD 0a and GSD 0b are due to insufficient glycogen storage, most GSDs are unable to remove glucose from glycogen (glycogenolysis), resulting in excess glycogen tissue storage. The first step in glycogenolysis is the release of glucose-1-phosphate (G-1-P) from glycogen by the action of glycogen phosphorylase. GSD type V is caused by mutations in the glycogen phosphorylase gene-specific for muscle (PYGM). Mutations in the glycogen phosphorylase gene specific for the liver (PYGL) cause GSD type VI.[4] The glucose-1-phosphate released by glycogen phosphorylase is then converted to glucose-6-phosphate (G-6-P) by the action of phosphoglucomutase. Glucose-6-phosphate in the liver is, in turn, converted to glucose by glucose-6-phosphatase encoded by the G6PC gene.[4] The resulting glucose is released into the blood as an energy source for other tissues/organs (Figure 2).
It should be noted that skeletal muscles lack glucose-6-phosphatase and therefore do not release glucose into the blood. GSDs type I results from genetic disorders in the metabolism of glucose-6-phosphatase.[5] GSD type Ia (also called von Gierke disease) is caused by mutations in the G6PC gene. Glucose-6-phosphate is synthesized in the cytoplasm of hepatocytes and must be transported into the lumen of the endoplasmic reticulum (ER), where it is acted upon by glucose-6-phosphatase yielding glucose, which is transported back to the cytoplasm and then through the hepatic GLUT2 transporter into the blood.[5]
Glucose-6-phosphate translocase1 (G6PT1) is the transporter protein that provides a G-6-P channel between the cytoplasm and the ER. The G6PT protein is made of three subunits termed G6PT1, G6PT2, and G6PT3 (Figure 2). Mutations in the SLC37A4 gene, which encodes the G6PT1 protein, are responsible for GSD type Ib (Figure 1). Fanconi-Bickel disease is a rare GSD caused by a GLUT2 deficiency due to a mutation in the SLC2A2 gene.[5] GLUT2 deficiency results in a failure to export glucose, an increased intracellular glucose level, and reduced glycogen degradation. This leads to increased glycogen storage and hepatomegaly.
As mentioned above, glycogen is a branched polymer. While glycogen phosphorylase works well at removing glucose from alpha-(1,4)-linkages, it does not work at branch points. Branch points are alpha-1,6 linkages. For removal of branch points, a glycogen debranching enzyme (GDE) is required, which in mammals is called “ammylo-alpha-1,6-glucosidase, 4-alpha-glucanotransferase” encoded by the gene AGL.[6] GSD type III is caused by mutations in the AGL gene, resulting in either a nonfunctional GDE enzyme (GSD type IIIa or type IIIb) or a GDE with reduced function (GSD type IIIc and IIId).[6]
GSD type II is unique among GSDs because it is also classified as a lysosomal storage disease (LSD).[7] Lysosomes are subcellular organelles that recycle cellular macromolecules. Lysosomal storage diseases are caused by a missing or nonfunctional lysosomal enzyme. In the case of GSD II, this enzyme is lysosomal acid alpha-glucosidase encoded by the gene GAA, which breaks down glycogen into glucose for use as a cellular energy source. Mutation in the GAA gene results in the toxic accumulation of glycogen in lysosomes.
Epidemiology
The true incidence of metabolic diseases is difficult to determine given the lack of uniform, universal screening at birth. Individual incidence of specific GSD types is further complicated due to overlap in symptoms and the lack of standardized specific testing in most areas of the world. A study evaluating the incidence of inborn errors of metabolism in British Columbia in the 1990s reported that the incidence of these diseases was approximately 30 cases per 100 000 live births.[8] This represented a mix of metabolic disorders; however, this was not restricted to glycogen storage diseases. Approximately 2.3 children per 100 000 births were thought to have glycogen storage disease in this study.[8] Current literature suggests the incidence of GSDs is approximately 1 case per 20000 to 43000 live births.[2]
Pathophysiology
As stated above, glycogen is the stored form of glucose and is composed of long polymers of 1,4 linked glucose with branch points via 1,6 linked glucose molecules.[4] The primary physiologic function of glycogen is to provide glucose via glycogenolysis for glucose homeostasis. The liver stores are used to maintain glucose homeostasis in the serum, and the muscle stores provide glucose for the muscles during periods of high demand/exercise as a source of energy.[4]
When these physiologic functions are defective, hypoglycemia, hepatomegaly, muscle cramps, exercise intolerance, and weakness develops. Some disorders also affect the myocardial tissue and can lead to cardiomyopathy and cardiac conduction defects.[9] Failure to maintain glucose homeostasis triggers alternate pathways to meet the metabolic demands. In GSD type 1, for example, failure of glycogenolysis in the liver results in increased lactic acid production (lactic acidosis) due to the intracellular accumulation of glucose-6-phosphate, which stimulates the glycolytic pathway.[5]
History and Physical
GSDs are a diverse set of rare inborn errors of carbohydrate metabolism that can have variable phenotypic presentation even within the same GSD type. Obtaining a family pedigree is useful in establishing the mode of inheritance. Most GSDs show an autosomal recessive inheritance, but a few (GSD type IX) show an x-linked inheritance.[5] Common presenting symptoms include growth retardation/poor weight gain in children, exercise intolerance, hypoglycemia, hepatomegaly, low muscle tone, acidosis, and hyperlipidemia.[2] As stated above, the most common presenting symptoms of these disorders are hypoglycemia and exercise intolerance.[2]
Patients with a defect in hepatic glycogen metabolism usually present with fasting hypoglycemia and ketosis. Their symptoms improve with glucose administration. Patients with a defect in skeletal muscle glycogen metabolism present with fatigue and exercise intolerance after short periods of moderate-intense exercise.[2] This is in contrast to those with a defect in fatty acid metabolism, as patients with defects in this pathway usually develop symptoms after prolonged exercise. Patients with skeletal muscle-associated GSD report muscle cramps and may present with rhabdomyolysis and/or myoglobinuria. In rare cases, progressive weakness may be reported. This, however, is usually limited to GSD type 0, II, and IV.[2]
In rare instances, GSD type III, V, and VII can present with weakness rather than muscle cramps and, over time, develop fixed weakness.[10] Patients with defects affecting cardiac muscles will present with symptoms of cardiomyopathy and rarely conduction defects.[9]
Anthropometric measurements should be obtained and graphed in all patients with GSDs to assess the overall growth pattern. Short stature or poor linear growth, especially in a child with hypoglycemia, should warrant workup for glycogen storage disorders.[2] The inability to properly release glucose from glycogen can result in the abnormal accumulation of glycogen. In the liver, this results in hepatomegaly with the potential for cirrhosis.[2]
Evaluation
Hypoglycemia is defined as a plasma concentration of glucose that results in symptoms attributable to hypoglycemia and is reversed with the administration of glucose. There is no set plasma glucose level above which GSDs can be ruled out, particularly for children. It is important to note that neonates go through a period of transitional hypoglycemia in the first 48 hours of life, during which GSDs cannot be diagnosed.[11]
Duration of fasting that leads to symptoms of hypoglycemia is an important element of history that must be obtained. A short duration of fasting that results in typical symptoms suggests glycogen storage disorder type I or III.[2] If an overnight duration of fasting leads to hypoglycemic symptoms, then glycogen storage disease type 0, VI, or IX should be considered.[2]
Laboratory Testing
Hypoglycemia should be documented by measuring serum glucose levels. In patients where hypoglycemia is suspected, a diagnostic fasting glucose test can be performed but should only be considered in a monitored inpatient setting.[12] Hepatic glycogen storage disorders (type 0, III, VI, and IX) are characterized by ketosis and usually yield a beta-hydroxybutyrate level greater than 2.5 mmol/L.[2] They will also typically present with hyperlipidemia and elevated liver function tests. Patients with glycogen storage disease type III also have elevated creatine kinase levels.[2] Glycogen storage disorder type I is associated with elevated levels of lactic acid and acidosis.[13]
Patients with type I disorder will also present with elevated liver enzyme and uric acid levels. Triglyceredemia is also common. Urinary myoglobin levels can be detected in patients with GSDs as well, particularly in those affected by GSDs that primarily affect the skeletal muscles.
Biopsy
Although specific genetic testing is now available for diagnosing most GSDs, histologic examination of liver or muscle biopsy is still used in specific scenarios. In GSD type 0, a liver biopsy will show decreased hepatic glycogen and can make a definitive diagnosis for this disease.[14] In GSD type I, a liver biopsy will reveal pale-staining, swollen hepatocytes, steatosis, and nuclear hyper-glycogenation.[2] Fibrosis is another common finding on liver biopsy in patients with GSDs and is predominant in patients with GSD type III, IV, and VI.[2] In GSD type III, periportal fibrosis and micronodular cirrhosis will be seen with distended hepatocytes due to excess glycogen accumulation.[15]
Muscle biopsies will reveal diastase-sensitive vacuoles and positive for periodic acid-Schiff (PAS) and acid phosphatase in GSD type IV.[16] In GSD type V, a muscle biopsy will reveal negative histochemical staining for phosphorylase activity.[17] In the absence of phosphorylase activity, the specimen remains brown instead of taking up a deep blue hue. In addition, the biopsy will reveal subsarcolemmal deposits of glycogen detected with periodic acid-Schiff (PAS) stain.[17] In patients with GSD type XV, a muscle biopsy will reveal PAS-positive inclusions that will not be digested with alpha-amylase treatment and, on electron microscopy, will be seen as filamentous material corresponding to polyglucosan bodies.[18]
Molecular Testing
Molecular genetic testing is noninvasive and, for the most part, available for diagnosing these rare genetic disorders. In some cases, they have eliminated the need for invasive muscle and liver biopsies. The genetic foci of mutations for these disorders are outlined in the following chart.[19][2][3]
| Glycogen synthase-2 deficiency (GSD type 0a) |
GYS2 |
| Glucose-6-phosphatase deficiency (GSD type Ia) |
G6Pase gene mutations or G6PC1 catalytic activity |
| Glucose-6-phosphate transporter deficiency (GSD type Ib) |
SLC37A4 |
| Glycogen debrancher deficiency (GSD type III) |
AGL |
| Glycogen branching enzyme deficiency (GSD type IV) |
GBE1 |
| Liver phosphorylase deficiency (GSD type VI) |
PYGL |
| Phosphorylase kinase deficiency (GSD type IXa) |
PHKA2 |
| GLUT2 deficiency or Fanconi-Bickel disease |
SLC2A2 |
| Muscle phosphorylase deficiency (GSD type V) |
PYGM |
| Phosphofructokinase deficiency (GSD type VII) |
PFKM |
| Phosphoglycerate mutase deficiency (GSD type X) |
PGAM |
| Lactate dehydrogenase A deficiency (GSD type XI) |
LDHA |
| Aldolase A deficiency (GSD type XII) |
ALDOA |
| Beta-enolase deficiency (GSD type XIII) |
ENO3 |
| Phosphoglucomutase-1 deficiency (GSD type XIV) |
PGM1 |
| Lysosomal acid maltase deficiency (GSD type IIa) |
GAA alpha-glucosidase enzyme (GAA) activity or GAA gene mutation |
| Lysosome-associated membrane protein 2 deficiency (GSD type IIb) |
LAMP2 |
| Glycogenin-1 deficiency (GSD type XV) |
GYG1 |
| Muscle glycogen synthase deficiency (GSD type 0b) |
GYS2 |
Treatment / Management
Currently, there is no cure for any GSD, and most treatments attempt to alleviate signs/symptoms. Key goals are to treat or avoid hypoglycemia, hyperlactatemia, hyperuricemia, and hyperlipidemia. Hypoglycemia is avoided by consuming starch, and an optimal, physically modified form is now commercially available. Hyperuricemia is treated with allopurinol and hyperlipidemia with statins. Some GSDs like GSD type II can now be treated with enzyme replacement therapy (ERT), using recombinant alglucosidase alfa, which degrades lysosomal glycogen.[20]
There is ongoing research to use ERT with other forms of GSDs. Liver transplantation should be considered for patients with certain GSDs with progressive hepatic forms that have progressed to hepatic malignancy or failure. Though liver failure and hypoglycemia may be corrected with liver transplantation, cardiomyopathy associated with the GSD will not be corrected and may continue to progress.[2]
Immediate management of acute hypoglycemia requires rapid correction with oral carbohydrates and/or parenteral glucose. Glucagon is only effective in insulin-mediated hypoglycemia and will not be helpful in patients who present with hypoglycemia secondary to a GSD.[21] A brief overview of therapeutic management for each of the GSDs is outlined in the following table.[2][3]
| GSD type 0, III, IV, VI, IXA, |
Uncooked or modified cornstarch to prevent hypoglycemia and liver failure in severe cases |
| GSD type Ia, Ib |
Uncooked or modified cornstarch for hypoglycemia, allopurinol for hyperuricemia, and liver transplantation in severe cases |
| GLUT2 deficiency or Fanconi-Bickel disease |
Frequent small meals to prevent hypoglycemia, uncooked or modified cornstarch, and restriction of galactose |
| Muscle phosphorylase deficiency (GSD type V) |
Glucose loading prior to exercise |
| Lysosomal acid maltase deficiency (GSD type IIa) |
Enzyme replacement therapy, modified cornstarch to prevent hypoglycemia, and liver transplantation if needed |
| GSD type IIb, VII, X, XI, XII, XIII, XIV, XV |
No specific treatment |
Differential Diagnosis
- Charcot-Marie-Tooth disease
- Congenital disorders of glycosylation
- Congenital lactic acidosis
- Deficiencies of phosphofructokinase
- Disorders of uric acid metabolism
- DNA depletion syndrome
- Duchenne muscular dystrophy
- Duchenne-Becker muscular dystrophy
- Endocardial fibroelastosis
- Fatty acid oxidation disorders
- Fructose-1-phosphate aldolase deficiency
- Fructose-1,6-biphosphatase deficiency
- Galactosemia
- Hyperlipoproteinemia
- Limb-girdle muscular dystrophy
- Limb-girdle muscular dystrophy
- Lysosome-associated membrane protein 2 deficiency
- Mitochondrial DNA depletion syndromes
- Mitochondrial myopathies
- Neurovisceral sphingolipidosis
- Niemann-Pick disease
- Organic acidurias
- Organic acidurias
- Polymyositis
- Sphingomyelinase deficiency
- Spinal muscular atrophy
- Werdnig-Hoffman disease
- Zellweger syndrome
Prognosis
With early diagnosis and proper management, the prognosis of most GSDs is good.[3] Good dietary management decreases the incidence of hypoglycemia-associated complications and has also been shown to improve renal dysfunction in patients with GSD type I.[2]
Rarely, end-stage renal disease requiring kidney transplantation may occur in patients with GSD type Ib.[22] In patients with GLUT2 deficiency, liver size and glycogen content significantly reduce after an antiketogenic diet.[23]
Complications
Hypoglycemia-associated seizures and cardiac arrest can occur in early childhood.[3] In patients with GSD type Ia, growth delay with short stature, renal dysfunction, hypertriglyceridemia, and hepatocellular carcinoma can occur. whereas in GSD type Ib, recurrent bacterial infections secondary to neutropenia will be seen.[3] In GSD type IV, progressive liver failure with cirrhosis can occur. Cardiomyopathy and limb-girdle dystrophy can be seen in patients with GSD type II. Hypertrophic cardiomyopathy is a classic complication of GSD type III.[3]
Growth retardation and short status are also seen in GSD type IX (a, b, c, d) and GSD type XII, but a cognitive-developmental delay is also a feature in the latter.[3] In GSD types V and XIII, exercise intolerance and rhabdomyolysis with an associated renal injury can occur.[3]
Deterrence and Patient Education
Patient and parent education about the dietary modifications and frequency of feeding is of utmost importance in optimizing the clinical outcomes for patients affected with these diseases. Depending on the type of GSD affecting the patient, specific instruction will be required. Patients and parents will need specific education to monitor for signs of hypoglycemia and the increased need for glucose or carbohydrate during an acute illness such as an infection.[2]
In patients with GLUT2 deficiency, additional education regarding oral replacement of electrolytes lost via the kidneys is essential.[23] These patients will also need dietary instructions to avoid galactose and follow a diet consisting of frequent small meals with adequate caloric intake to ensure optimal growth.[23]
Enhancing Healthcare Team Outcomes
GSDs are a group of complex metabolic disorders best managed by an interprofessional team of clinicians, nurses, pharmacists, and dietitians. At present, there is no cure for any GSD, and most treatments attempt to alleviate signs/symptoms. However, even this symptomatic management requires vigorous patient/parent education to ensure dietary restrictions and frequency of carbohydrate administration are appropriately managed.
Registered dieticians and specialty nurses play a key role in educating patients and their caregivers to ensure hypoglycemia is avoided. This not only ameliorates the risk of hypoglycemia-associated complications but also prevents long-term disease sequelae in most GSDs. Specialty pharmacists play a pivotal role in managing GSD type II to ensure enzyme replacement therapy is carried out adequately and that the medication is administered under optimal circumstances. Primary care clinicians, which include physicians and mid-level practitioners, and pediatricians, in coordination with specialists, help ensure patients have adequate growth and function with minimal risk of severe complications such as renal or liver failure.
All interprofessional team members should be vigilant in monitoring these patients and alert the other team embers if any complications develop or the patient's condition worsens; this requires meticulous documentation and open communication between everyone on the care team. The key overall goal is to avoid and treat hypoglycemia, hyperlactatemia, hyperuricemia, and hyperlipidemia. A well-coordinated interprofessional team can help manage patients with these diseases adequately and ensure they live a normal life. [Level 5]
The development of experimental therapies, such as gene therapy, may eventually provide curative options for patients with these diseases in the future.[24]