Continuing Education Activity
Acute febrile neutrophilic dermatosis (AFND), also known as Sweet syndrome or Gomm-Button disease, manifests as erythematous plaques and papules, coupled with constitutional symptoms like fever and malaise. With an average onset age of 30 to 60 years and a female predominance, AFND displays a dense neutrophilic infiltrate histologically. Linked with various conditions, including infections, malignancies, inflammatory bowel disease, autoimmune disorders, pregnancy, and drugs, its cutaneous lesions present as tender, edematous, erythematous plaques, and occasionally bullous. Prednisone stands as the primary treatment, although dapsone, colchicine, and potassium iodide present as potential alternatives. This activity offers insights into AFND evaluation and management strategies and emphasizes the interprofessional team's role in optimizing patient care.
Objectives:
Identify the etiology of acute febrile neutrophilic dermatosis.
Apply appropriate evaluation methods for acute febrile neutrophilic dermatosis.
Implement evidence-based treatment strategies for acute febrile neutrophilic dermatosis.
Develop interprofessional team strategies for improving care coordination and communication to advance the treatment of acute febrile neutrophilic dermatosis and improve outcomes.
Introduction
Acute febrile neutrophilic dermatosis (AFND), also known as Sweet syndrome or Gomm Button disease, consists of erythematous plaques and papules along with constitutional symptoms such as fever and malaise. The average age of onset is 30 to 60 years, and there is a female predominance. The disease is characterized histologically by a dense neutrophilic infiltrate. It is associated with several underlying diseases, including infections, malignancies, inflammatory bowel disease, autoimmune disorders, pregnancy, and drugs. Clinically, the cutaneous lesions present as tender, edematous, erythematous plaques and papules, which may occasionally be bullous. The most effective treatment is prednisone. Other potential treatments include dapsone, colchicine, and potassium iodide.[1]
Etiology
Etiologically, acute febrile neutrophilic dermatosis can be classical or idiopathic, malignancy-associated, or drug-induced. Each of these ultimately leads to either unsolicited neutrophil proliferation or persistence in the blood and tissue, which is responsible for the clinical manifestations of the disease.
Classical
Classical Sweet syndrome, also known as idiopathic Sweet syndrome, accounts for the majority of Sweet syndrome cases. It is characterized by meeting the recognized diagnostic criteria without association with cancer or drug exposure.[1] The occurrence of classical Sweet syndrome can be observed in various medical circumstances.
Infections, including those affecting the upper respiratory tract and gastrointestinal system, have been found to be associated with the development of Sweet syndrome. Typically, this syndrome manifests within 1 to 3 weeks following the initial infection. This syndrome has also been associated with inflammatory bowel disease (Crohn disease and ulcerative colitis) and pregnancy. There are infrequent and less conclusive connections observed with various infections such as HIV, tuberculosis, chlamydia, and viral hepatitis, as well as primary immunodeficiencies, autoimmune disorders including Behçet syndrome, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, autoimmune thyroid disease, connective tissue disorders such as systemic lupus erythematosus and dermatomyositis, and vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome.[2]
VEXAS syndrome is an autoinflammatory condition that has been documented in adult males. Dermatological manifestations were observed in a significant proportion of patients diagnosed with VEXAS syndrome, as evidenced by a study conducted in 2022 that reviewed the literature and identified 126 out of 141 patients (89%) exhibiting such manifestations. The most commonly observed cutaneous manifestation appears to be the presence of lesions resembling those seen in Sweet syndrome, characterized by painful, erythematous, or violaceous nodules. Additional dermatological characteristics encompass the presence of cartilaginous engagement, including auditory and nasal chondritis, cutaneous vasculitis, and periorbital angioedema.[3]
Malignancy-associated
Malignancy-associated Sweet syndrome is responsible for a considerable proportion of instances of Sweet syndrome.[4] The occurrence of malignancy-associated Sweet syndrome in children seems to be predominantly observed in individuals aged 3 years and older.[5] Infants have been found to have rare cases of Sweet syndrome linked to cancer.[6] The occurrence of Sweet syndrome can manifest prior to, subsequent to, or simultaneously with the development of a tumor.[7] The occurrence of Sweet syndrome in individuals with a prior history of cancer may indicate the potential for disease relapse.[1] The occurrence of Sweet syndrome is more commonly observed in conjunction with hematologic malignancies as opposed to solid tumors.[8]
Acute myeloid leukemia (AML) is the neoplastic condition that exhibits the highest frequency of association with Sweet syndrome. AML represents 42% of the hematologic malignancies, whereas myeloproliferative disorders were identified as the second most prevalent malignancy category, accounting for 22%.[9] Cancers of the genitourinary tract, breast, and digestive tract are the solid tumors most often linked to Sweet syndrome.[10] Previous research conducted on individuals diagnosed with Sweet syndrome has revealed that those who are older, experience anemia, thrombocytopenia, and leukopenia, and do not exhibit arthralgias are more prone to developing malignancy-associated Sweet syndrome.[7][8] Furthermore, it is worth noting that there is an increased risk of malignancy in patients who exhibit the histiocytoid or subcutaneous histologic variation of Sweet syndrome.[11]
Drug-induced
Multiple drugs may contribute to Sweet syndrome. Granulocyte-colony stimulating factor (G-CSF) is the most widely reported contributory medication.[12] It has been documented that checkpoint inhibitor treatment can cause neutrophilic dermatoses, such as Sweet syndrome, brought on by ipilimumab therapy.[13] Sweet syndrome often manifests around two weeks following drug exposure in individuals with no previous record of exposure to the causative substance.[14] Reexposure to the initiating substance frequently causes a recurrence.[1] Anecdotal case reports suggest an association with antibiotics, antiepileptics, nonsteroidal antiinflammatories, retinoids, and diuretics. However, the causal association has not been established by rechallenge.[1][15]
Epidemiology
AFND is uncommon, found worldwide, and has a 4:1 female-to-male predominance. The average age of onset is 30 to 60 years, but it can occur in infants and older people. The observed gender disparity in Sweet syndrome does not seem to apply to the pediatric population.[5] Sweet syndrome does not appear to have a clear racial preference. At least half of all patients have an identifiable underlying association. Internal malignancy is found in 15% to 30% of cases, and hematologic malignancy is more common than solid organ cancer. Twenty-five percent of patients have a preceding infection, and 10% have exposure to a potentially causative drug.[16]
Pathophysiology
The underlying pathophysiology of acute febrile neutrophilic dermatosis is unknown, but a few hypotheses exist. The etiology of this condition has been postulated to involve many factors, such as hypersensitivity reactions, dysregulation of cytokines, and hereditary predisposition.
Hypersensitivity Reaction
An immunological response to bacterial, viral, neoplastic, or other antigens can influence the development of Sweet syndrome. This immune response stimulates the generation of cytokines that facilitate neutrophil activation and infiltration. The notion is somewhat supported by the good response observed in systemic glucocorticoid treatment and the reported improvement of Sweet syndrome after treating bacterial infections or malignancies.[17]
Cytokine Dysregulation
Dysregulation of cytokine secretion (including interleukin-1 [IL-1], G-CSF, granulocyte-macrophage colony-stimulating factor, and interferon-gamma) resulting in dysfunction of neutrophil apoptosis is one hypothesis.[18] A defect in the transcriptional regulation of the hematopoietic protein tyrosine phosphatase nonreceptor type 6 (PTPN6) has also been found to contribute to the pathogenesis of neutrophilic dermatoses.[19] Alterations of proinflammatory cell signaling pathways and cytokine production in a genetically predisposed individual are considered central to the pathogenesis of AFND. Ultimately, these alterations affect neutrophil proliferation, migration, and destruction in the skin and other tissues.
Tissue neutrophil levels are a function of infectious triggers for neutrophil proliferation and migration, bone marrow granulopoiesis, storage and release, transendothelial migration, and the rate of neutrophil destruction. Abnormality in any of these steps could influence the development and progression of AFND. Migration of neutrophils to the tissue in situations of cell stress is a normal immunological response; however, the loss of control over this process in AFND appears to be largely cytokine-mediated. Most commonly, IL-1, IL-3, IL-6, and IL-8 are elevated in AFND. Moreover, elevated levels of G-CSF have also been noted in patients with AFND.
Various tissues, including the endothelium and macrophages, produce G-CSF. Additionally, increased production of G-CSF is noted in multiple malignancies. In sum, it may be hypothesized that abnormally increased production of G-CSF or increased sensitivity to normal levels of G-CSF could be the unifying mechanism of all three variants of AFND.
Genetic Factors
Defects in the transcriptional regulation of the PTPN6 gene and mutations of the MEFV gene associated with familial Mediterranean fever have been implicated in the pathogenesis of AFND.[1][20][21] The association between HLA-B54 and Sweet syndrome has been observed in Japanese patients.[22]
Antibodies against neutrophilic cytoplasmic antigens have been seen in certain individuals diagnosed with Sweet syndrome.[23] Nevertheless, the potential significance of these antibodies in causing disease has yet to be determined. Additional research is required to elucidate the underlying mechanisms responsible for the pathogenesis of Sweet syndrome.
Histopathology
A 4-mm punch biopsy is usually enough to produce a tissue specimen for histologic evaluation in patients with inflammatory papules or plaques. Microbial pathology stains should be performed since the infection may resemble Sweet syndrome. In addition, a second 4-mm punch biopsy is obtained for bacterial, fungal, and mycobacterial cultures. A swab specimen can also be sent for culture if pustules are detected. An excisional biopsy is preferred in patients with nodules suggestive of subcutaneous Sweet syndrome (a variant in which the pathologic process is primarily located in the subcutaneous fat) because it provides a more generous sample of the subcutaneous fat, which may aid in the interpretation of the histologic findings.
The histopathological features usually depend upon the type of lesion sampled:
- Epidermis: Usually insignificant; however, the occasional spillover of dermal neutrophils may occur, producing subcorneal pustules. There can be epidermal spongiosis. Sometimes, reticular degeneration of the keratinocytes may lead to frank intraepidermal or subepidermal vesiculation.
- Dermis: Diffuse nodular and perivascular neutrophilic infiltrate without evidence of vasculitis; leukocytoclasia, with endothelial swelling without fibrinoid necrosis, is a usual finding. Vasculitis may be seen as a secondary epiphenomenon in this setting.
- Subcutaneous fat: Extension of dermal infiltrate into the subcutis may lead to septal or lobular panniculitis. Isolated neutrophilic panniculitis has also been observed.
Histopathological variants:
- Histocytoid: An infiltrate composed of immature spindle-shaped histiocyte-like cells. Immunohistochemistry shows the presence of myeloperoxidase activity in these cells. They have to be closely differentiated from leukemia cutis.
- Lymphocytic: The infiltrate is predominantly lymphocytic, usually associated with myelodysplasia.
- Eosinophilic: The infiltrate is rich with eosinophils. A case has been observed in the setting of enteropathy-associated T-cell lymphoma.[1][11]
History and Physical
The term AFND is used to describe the occurrence of temperatures over 38°C in individuals with Sweet syndrome. The majority of patients diagnosed with drug-induced Sweet syndrome often experience fever. In contrast, there is a subset of individuals with classical or malignancy-associated disease where fever may be absent in 10% to 20% of cases. Additional symptoms commonly observed in Sweet syndrome include arthralgias, malaise, headaches, and myalgias. Alongside the sudden appearance of the painful inflammatory papules, plaques, and nodules that are characteristic of Sweet syndrome, there may be the emergence of additional clinical characteristics:
Cutaneous Disease
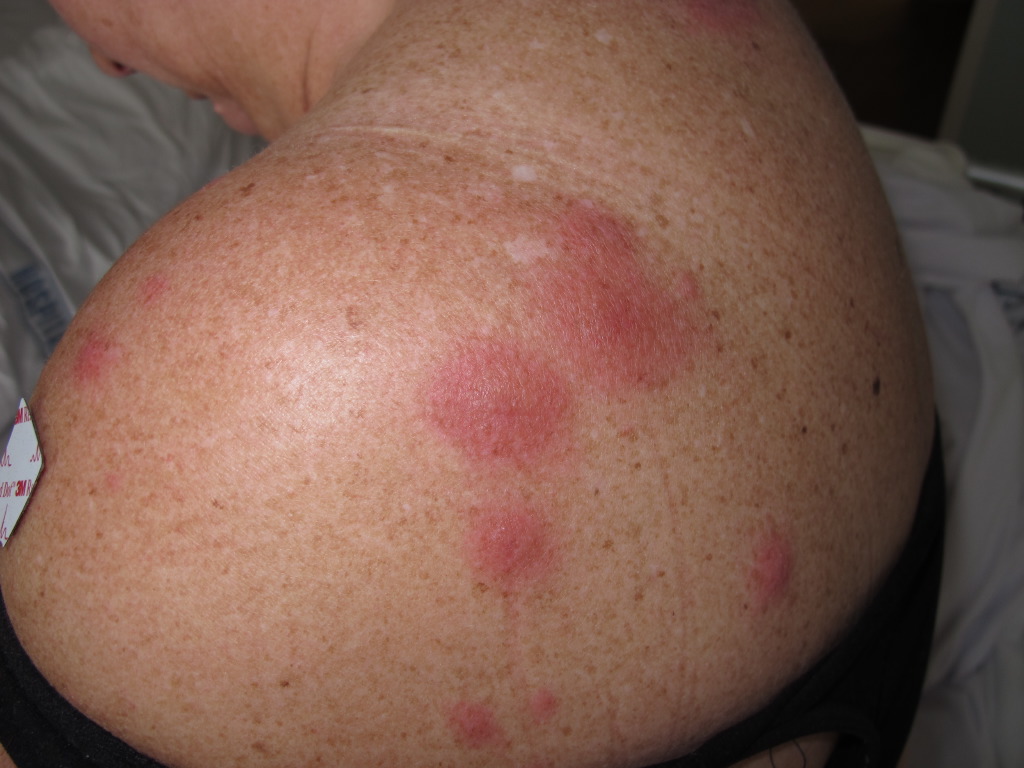
The classical clinical appearance is erythematous, tender, nonpruritic papules, nodules, and plaques with predominantly head, neck, and upper trunk distribution patterns. The plaques are often studded with multiple closely grouped translucent papules, giving the illusion of vesiculation. These pseudo-vesicles and pseudo-pustules impart a mamillated appearance to the surface of the plaques and are considered one of the suggestive manifestations of AFND. Subsequent ulceration may occur, which resembles a superficial pyoderma gangrenosum described in association with acute myeloid leukemia. Papulonodular lesions of the lower limbs closely resemble erythema nodosum and can be differentiated histopathologically. Sweet syndrome patients report soreness or burning from cutaneous lesions.
Less often observed manifestations of Sweet syndrome include:
- Neutrophilic dermatosis of the dorsal hands: Characterized by bluish violaceous or hemorrhagic papules, bullae, and nodules on the dorsal hands. It may be accompanied by typical lesions of Sweet syndrome in other areas of the body. This variant may have a more significant association with occult or hematological malignancies. It closely mimics a bullous pyoderma gangrenosum.
- Subcutaneous Sweet syndrome: Presents with erythema nodosum-like lesions on the shins. Notably, the location of neutrophilic infiltration occurs inside the subcutaneous adipose tissue, as opposed to the dermis.[1][24]
- Bullous Sweet syndrome: Characterized by vesicles and flaccid bullae, Bullous Sweet syndrome is rare and can cause pyoderma gangrenosum-like ulceration, usually in hematologic malignancies.[4]
Furthermore, there have been documented cases of patients presenting with Sweet syndrome that closely resemble necrotizing fasciitis, referred to as necrotizing Sweet syndrome[25]. Patients exhibit pyrexia, accompanied by the progressive development of erythematous to violaceous plaques. The manifestation of extensive cellulitis-like plaques has been identified as an infrequent clinical sign of Sweet syndrome. The designation "giant cellulitis-like Sweet syndrome" was employed to describe the occurrence of extensive infiltrating inflammatory plaques and bullae in three patients who met the diagnostic criteria for Sweet syndrome.[26] The phenomenon of pathergy, wherein a cutaneous lesion is triggered by minor trauma, is demonstrated in AFND in common with other neutrophilic dermatoses. The probable mechanism could be the destruction of endothelial cells, cytokine dysregulation, and resultant neutrophil activation.
Oral Disease
The occurrence of oral manifestations in classical Sweet syndrome is infrequent. Nevertheless, it is worth noting that around 12% of individuals diagnosed with Sweet syndrome in connection with hematologic malignancies experience the development of oral ulcers as a manifestation of the disease, specifically on the buccal mucosa or tongue.[1] Further documented oral observations encompass bullae, vesicles, gingival hyperplasia, necrotizing ulcerative periodontitis, nodules, papules, pustules, and swelling of the tongue.[27]
Extracutaneous Disease
Sweet syndrome is characterized by the potential for neutrophilic infiltration into various organ systems, including the eye, lungs, muscles, bone, liver, spleen, heart, kidneys, central nervous system, and gastrointestinal tract. Such infiltration may give rise to symptoms particular to the extracutaneous site of inflammation.
- Eye: Ocular inflammation is a prevalent extracutaneous presentation, manifesting in 17% to 72% of patients diagnosed with the classical variant. Conjunctivitis, episcleritis, scleritis, limbal nodules, peripheral ulcerative keratitis, iritis, glaucoma, dacryoadenitis, and choroiditis are all illustrative instances of ocular manifestations.[28]
- Musculoskeletal system: An additional common site of extracutaneous involvement in the body, where arthralgias, arthritis, and myalgias can manifest.
- Central nervous system: Encephalitis, aseptic meningitis
- Pulmonary system: Neutrophilic alveolitis, pleural effusions, airway obstruction
- Cardiovascular system: Myocarditis, aortitis and aortic stenosis, coronary artery occlusion
- Liver: Hepatitis, hepatomegaly
- Intestines: Neutrophilic inflammation of the intestines
- Spleen: Splenomegaly
- Kidneys: Mesangial glomerulonephritis, hematuria, proteinuria
- Bone: Sterile osteomyelitis
Additionally, there have been reports of the occurrence of systemic inflammatory response syndrome (SIRS) in conjunction with Sweet syndrome.[29]
Evaluation
There are no specific diagnostic tests for AFND. Su and Liu proposed the diagnostic criteria for Sweet syndrome.[30]
Major Criteria
Both of the following must be present to establish a diagnosis of Sweet syndrome:
- Abrupt onset of typical cutaneous lesions
- Histopathology consistent with Sweet syndrome
Minor Criteria
For diagnostic confirmation of Sweet syndrome, at least 2 minor clinical features should be present.
- Preceded by one of the associated infections or vaccinations, accompanied by one of the associated malignancies, drug exposure, or pregnancy
- Fever >38º C
- Three out of these 4 abnormal laboratory values at presentation, including 1) Erythrocyte sedimentation rate (ESR) >20 mm/h; 2) Leukocytes >8000 mm3/h; 3) Neutrophils >70%; 4) Elevated C-reactive protein (CRP)
- Dramatic response to systemic corticosteroids or potassium iodide (KI)
Based on these criteria, the essential investigations include a skin biopsy, complete blood count, ESR, CRP, liver, and renal function tests. Additional tests include thyroid function tests, rheumatoid factor, and antistreptolysin-O antibody titers. The most prevalent laboratory abnormality in Sweet syndrome patients is peripheral leukocytosis with neutrophilia.
Neutrophilia is found in the great majority of patients with classical Sweet syndrome, as well as in a considerable proportion of patients with cancer-related and drug-induced illnesses. Anemia and platelet abnormalities are common in patients with Sweet syndrome caused by cancer or medications, but they are seldom found in patients with classic Sweet syndrome. Abnormalities in the full metabolic panel and urine may indicate hepatic or renal involvement.[16]
A complete lymph node examination, examination of breasts and pelvis in females and prostate and testicles in males, sigmoidoscopy, and positron emission tomography (PET) scans comprise the workup to rule out occult malignancy. Malignancy testing should be considered only when there is a reasonable clinical suspicion for an underlying malignancy (eg, constitutional symptoms such as weight loss) and no other explanation for a Sweet syndrome diagnosis (eg, pregnancy, drug exposure, recent infection, inflammatory bowel disease, rheumatoid arthritis). Although elevated levels of anti-neutrophil cytoplasmic antibodies (C-ANCA) have been noted, they are no longer considered significant.[1][30]
Treatment / Management
To achieve illness control, the majority of patients require systemic medication. Patients who appear with a few isolated skin lesions (eg, <5% of body surface area) and no systemic symptoms, on the other hand, may react well to local corticosteroid therapy. In individuals with broad cutaneous lesions, local treatment may be utilized in addition to systemic glucocorticoids or other systemic medicines. This is frequently conducted to lessen reliance on systemic medications and speed up lesion healing.
Systemic Glucocorticoids
The immediate and dramatic response of Sweet syndrome to systemic glucocorticoids, as recorded in several case reports and case series, validates their place as the therapy of choice for this condition. Including the response to systemic glucocorticoids in the diagnostic criteria reflects the high chance of therapeutic effectiveness. Systemic glucocorticoids are beneficial for both cutaneous and extracutaneous Sweet syndrome signs. Adults are often started on oral prednisone at a dose of 0.5 to 1 mg/kg per day. Symptoms typically recover after 48 hours, and skin lesions disappear within 1 to 2 weeks.[31] Once disease control is achieved, tapering prednisone over a 4- to 6-week period should be attempted.
Topical and Intralesional Corticosteroids
Topical corticosteroids with high potency (eg, Clobetasol 0.05% ointment) and intralesional corticosteroid injections are used. For 2 to 3 weeks, topical corticosteroids are typically used twice daily on the afflicted area. Occlusion therapy may hasten the response to therapy. Intralesional therapy is effective for thick plaques on the trunk or extremities that do not respond entirely to topical therapy.
Colchicine, Dapsone, and Potassium Iodide
These medicines, less commonly used than systemic glucocorticoids, are utilized as first-line therapeutics when it is preferable to avoid systemic glucocorticoid therapy due to patient comorbidities or other circumstances. Treatment with low-dose steroids may have to be continued for a longer time to prevent exacerbations. If remission is not achieved within 3 months, a second-line steroid-sparing agent must be added.
The main second-line steroid-sparing agents include dapsone (50-100 mg/day), KI (300 mg 3 times a day), and colchicine (0.5 mg 3 times a day). A baseline G6PD activity should be done in patients before starting dapsone. Miscellaneous drugs that have been reported to be efficacious in various studies include cyclophosphamide, cyclosporine, thalidomide, clofazimine, intravenous immunoglobulin, and tumor necrosis factor–α antagonists.[1]
Clinical evidence suggests that pulse glucocorticoid therapy may be useful in treating severe Sweet syndrome or Sweet syndrome that does not respond adequately to other medications.[32] Adults may be given intravenous methylprednisolone at doses ranging from 500 to 1000 mg per day for 3 to 5 days. An oral glucocorticoid taper or another systemic immunosuppressive medication is used after treatment. In refractory instances, newer case reports suggest that rituximab, adalimumab, infliximab, tocilizumab, ustekinumab, and the Janus kinase (JAK) inhibitor baricitinib may be beneficial.[33][34][35][36][37]
Differential Diagnosis
Infection is one of the most significant illnesses to consider in a patient with Sweet syndrome–like clinical or histologic findings. As with Sweet syndrome, bacterial sepsis can cause fever, leukocytosis, and skin lesions with neutrophil-dense infiltrates. Furthermore, Sweet syndrome lesions can resemble local bacterial, fungal, or atypical mycobacterial diseases.
The results of a retrospective study in which fluorescence in situ hybridization (FISH) was performed on preserved cutaneous specimens from 5 of 6 patients with hematologic malignancy–associated histiocytoid Sweet syndrome suggest that the clinicopathologic findings of histiocytoid Sweet syndrome may be particularly difficult to distinguish from leukemia cutis.[38] Cytogenic abnormalities in lesional skin matched those found in earlier bone marrow biopsy specimens in 4 of the 5 patients, implying that these 4 individuals had leukemia cutis. More research is needed to understand the usefulness of FISH analysis in distinguishing between histiocytoid Sweet syndrome and leukemia cutis.
The clinical differential diagnosis for Sweet syndrome is based on lesion morphology. Data from the clinical history, physical examination, pathology findings, and microbiological investigations might help differentiate Sweet syndrome from other conditions, including:
- Erythematous, edematous plaques: Cutaneous infection (bacterial, fungal, and mycobacterial); urticaria and urticarial vasculitis; drug eruptions; halogenoderma (eg, bromoderma, iododerma); other neutrophilic dermatoses (eg, pyoderma gangrenosum, neutrophilic eccrine hidradenitis, Behçet syndrome, cutaneous metastatic Crohn disease)
- Bullous lesions: Bullous leukocytoclastic vasculitis; bullous pyoderma gangrenosum; autoimmune bullous diseases (eg, bullous pemphigoid, bullous systemic lupus erythematosus, inflammatory epidermolysis bullosa acquisita, linear immunoglobulin A bullous dermatosis); infection with bullous and hemorrhagic or necrotic changes (eg, bullous cellulitis, vessel invasive infections such as aspergillosis, ecthyma gangrenosum)
- Nodules (including subcutaneous Sweet syndrome): Cutaneous infection (eg, deep fungal infection, Majocchi's granuloma, atypical mycobacterial infection); subcutaneous sarcoidosis; erythema nodosum; vasculitis (particularly medium-vessel vasculitis such as cutaneous polyarteritis nodosa); malignancy (eg, lymphoma cutis, leukemia cutis, metastatic carcinoma)[1][21][39][40]
Prognosis
The cutaneous lesions of AFND, or Sweet syndrome, eventually show spontaneous resolution along with the associated extracutaneous manifestations. However, periodic exacerbations may occur, representing underlying paraneoplastic conditions. In a patient with a treated malignancy, the recurrence of lesions in Sweet syndrome heralds a relapse. In the case of local cutaneous complications such as scarring and acquired cutis laxa, the prognosis is poor as they seldom respond to any modality.[41]
In about 30% of cases, the skin lesions show spontaneous resolution within 3 months. However, a fluctuating course with relapse and remission is more frequently observed. Relapse can occur after reducing or discontinuing systemic glucocorticoids or other therapy, and it is more likely in patients with malignancy-related illnesses. Sweet syndrome recurrences occur in around 30% of patients with classical Sweet syndrome. In the presence of an underlying hematologic malignancy, the recurrence rate may be as high as 69%.[42]
Complications
Even though lesions usually resolve with mild post-inflammatory hyperpigmentation, various localized complications, such as scarring in the case of ulcerative lesions and atrophy in cases of panniculitis, have been observed. Postinflammatory elastolysis presenting as acquired cutis laxa, also called Marshal syndrome, is a frequently encountered finding.[41]
Deterrence and Patient Education
In patients diagnosed with AFND or Sweet syndrome, detailed counseling regarding the need to rule out underlying systemic diseases or malignancies should be carried out. Regular consultation with the dermatologist is recommended. In patients with treated malignancy, the reappearance of lesions should not be ignored, as they can signal a relapse.[24]
Enhancing Healthcare Team Outcomes
Sweet syndrome is one of the great mimickers in dermatology. The key to diagnosing this syndrome is to have a high index of suspicion. Prompt referral to a dermatologist for tender erythematous papulonodular lesions with fever, which show an inadequate therapeutic response to analgesics and antibiotics, is necessary. In suspected cases, primary clinicians should obtain routine investigations such as a complete blood count, ESR, and CRP. Sweet syndrome heralds an underlying infectious, immunological, or malignant disease; hence, prompt diagnosis and a thorough evaluation by an interprofessional team are necessary for ruling out underlying systemic illnesses.[1][24]