Continuing Education Activity
Systemic sclerosis or scleroderma is a rare connective tissue disorder with an unknown and complex pathogenesis. Scleroderma can be divided into 2 primary forms—localized scleroderma (including morphea, linear scleroderma, and scleroderma en coup de sabre) and systemic sclerosis. Systemic sclerosis can be further classified as limited systemic sclerosis (formerly known as CREST syndrome, characterized by calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) or diffuse systemic sclerosis based on clinical and serological criteria. Localized scleroderma primarily affects the skin and subcutaneous tissue, whereas systemic sclerosis is associated with systemic manifestations and internal organ involvement, leading to increased mortality. The manifestations of scleroderma may overlap extensively with those of other rheumatological or immunological diseases. The severity of the presentation may also vary depending on the timing of the systemic sclerosis diagnosis.
Systemic sclerosis affects multiple organ systems, necessitating a collaborative healthcare team involving primary care clinicians, rheumatologists, gastroenterologists, cardiologists, pulmonologists, nephrologists, and dermatologists to address the condition. Effective management of systemic sclerosis requires early diagnosis and ongoing disease progression monitoring. Although a definitive cure does not exist for systemic sclerosis, treatment primarily focuses on managing affected organs and alleviating symptoms to prevent further organ damage in individuals with systemic sclerosis. This activity addresses the presentation, evaluation, and management of systemic sclerosis and examines the role of an interprofessional healthcare team in caring for affected patients. This activity helps participating clinicians differentiate this tissue disorder from related rheumatological diseases.
Objectives:
Identify early clinical manifestations and risk factors associated with systemic sclerosis to facilitate prompt diagnosis and intervention.
Implement regular screening protocols for systemic sclerosis complications, including pulmonary function tests, echocardiography, and renal function assessments.
Apply evidence-based treatment strategies for systemic sclerosis, considering a patient's clinical presentation, severity, and comorbidities.
Collaborate with interprofessional healthcare team members to coordinate care, optimize treatment outcomes, and address the holistic needs of patients with systemic sclerosis.
Introduction
Systemic sclerosis, also known as scleroderma, is a rare connective tissue disorder with an unknown and complex pathogenesis. Scleroderma can be divided into 2 primary forms—localized scleroderma (including morphea, linear scleroderma, and scleroderma en coup de sabre) and systemic sclerosis. Systemic sclerosis can be further classified as limited systemic sclerosis (formerly known as CREST syndrome, characterized by calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) or diffuse systemic sclerosis based on clinical and serological criteria. Although significant progress has been made in understanding the pathophysiology of scleroderma over the past centuries, the disease continues to pose significant morbidity and mortality in patients.[1][2]
Localized scleroderma primarily affects the skin and subcutaneous tissue, leading to patches of thickened skin that, on biopsy, reveal dermal fibrosis similar to the histopathological changes seen in the thickened skin in systemic sclerosis. However, it is not associated with the Raynaud phenomenon, digital ischemic events, or internal organ involvement. Antinuclear antibodies may be present in up to 50% of cases of localized scleroderma; however, more specific autoantibodies such as anti-centromere, anti-Scl-70, and anti-RNA polymerase III are absent in this condition.[3] Notably, localized scleroderma is not associated with increased mortality. On the other hand, systemic sclerosis is associated with several systemic manifestations and internal organ involvement, leading to increased mortality, and its classification is based on skin involvement.[4]
Limited cutaneous systemic sclerosis, previously known as CREST syndrome, is characterized by skin thickening distal to the elbows and knees and/or on the face without trunk involvement. On the other hand, diffuse cutaneous systemic sclerosis involves skin thickening that may affect areas proximal to the elbows, knees, face, and/or trunk. Both limited cutaneous systemic sclerosis and diffuse cutaneous systemic sclerosis are associated with several systemic manifestations and positive autoantibodies. Antinuclear antibodies may be present in more than 90% of cases of systemic sclerosis, and up to 70% of cases exhibit at least one of the more specific autoantibodies (anti-centromere, anti-Scl-70, and anti-RNA polymerase III). Scleroderma most commonly affects the skin, gastrointestinal tract, lungs, kidneys, skeletal muscle, and pericardium among affected organs.[5] The manifestations of scleroderma may overlap extensively with those of other rheumatological or immunological diseases. The severity of the presentation may also vary depending on the timing of the systemic sclerosis diagnosis.
Etiology
The exact etiology of systemic sclerosis is not completely understood, and it is believed that both genetic and environmental factors are thought to contribute to its development.
Genetic Factors
Systemic sclerosis shows familial clustering and is associated with clusters of multiple other autoimmune diseases within families.[6][7] Genomewide association studies have confirmed the involvement of the major histocompatibility complex genetic region in systemic sclerosis, akin to other autoimmune disorders like systemic lupus erythematosus and rheumatoid arthritis. The association of specific human leukocyte antigens (HLA), including HLA DRB1*1104, DQA1*0501, and DQB1*0301, with systemic sclerosis has been long known. Further, non-HLA loci such as PTPN22, NLRP1, STAT4, and IRF5 have also been implicated in the etiology of systemic sclerosis.[8]
Environmental Factors
Several environmental triggers are associated with the subsequent development of systemic sclerosis, including infectious agents such as Cytomegalovirus (CMV), Epstein-Barr virus, and parvovirus B19.[9] Exposure to silica dust is also associated with systemic sclerosis, along with occasional exposure to other agents such as organic solvents, toluene, xylene, trichloroethylene, and polyvinyl chloride. Notably, cigarette smoking is not a proven risk factor for systemic sclerosis.
Several scleroderma-like disorders, which can be distinguished from systemic sclerosis by clinical, histopathological, and laboratory features, are associated with environmental exposures such as contaminated rapeseed cooking oil causing toxic oil syndrome and L-tryptophan linked to eosinophilia-myalgia syndrome.[10] In addition, certain drugs, such as bleomycin and cocaine, have been associated with developing systemic sclerosis-like illnesses.
Epidemiology
The rarity of systemic sclerosis contributes to sparse epidemiological data, with reported incidence and prevalence rates influenced by factors such as geography, case definition, and ascertainment methods. Overall, prevalence rates range between 38 and 341 cases per million persons, while incidence rates vary from 8 to 56 new cases per million persons annually globally.[11]
A study in the United States reported an estimated incidence of 19.3 new cases per million adults per year, with a prevalence of 242 cases per million adults in the Detroit area from 1989 to 1991.[12] Another study from Quebec in 2003 found a prevalence of 443 cases per million adults.[13] Interestingly, prevalence rates are higher in the United States and Australia compared to Europe and Asia (specifically, Japan and Taiwan).[14][15] Notably, the Choctaw Native Americans from Oklahoma exhibited the highest prevalence of systemic sclerosis at 660 cases per million adults, highlighting geographical variations in the prevalence of this condition.[16]
Systemic sclerosis exhibits a female predominance with a female-to-male ratio of about 5:1. Females typically develop the condition at an earlier age than males. The peak age of onset is between 45 and 54 in African-American females and between 55 and 64 in European-American females. Notably, systemic sclerosis is rare among children and teenagers aged 15 or younger.[17] Disease onset in individuals aged between 15 and 24 is also rare, with an incidence of 21.2 per million in African-American females and 11.16 per million in European-American females. Patients with African ancestry face a higher risk of developing systemic sclerosis, experiencing an earlier onset of the disease, and often developing more severe manifestations.[18]
Pathophysiology
The pathophysiology of systemic sclerosis is intricate and not entirely understood. The disease is characterized by 3 main hallmarks—vascular insult, autoimmunity, and tissue fibrosis. Different clinical phenotypes of systemic sclerosis are attributed to variations in the contribution of these factors in each patient's disease pathogenesis. Various triggers, such as viruses, environmental exposures, autoantibodies, proteolytic enzymes, and inflammatory cytokines, can initiate the initial vascular insult in systemic sclerosis. This initial vascular insult leads to endothelial cell activation, followed by overexpression of adhesion molecules, leading to the activation of platelets and thrombotic and fibrinolytic cascades.
Activated endothelial cells also release ET-1, a potent vasoconstrictor, and promote leukocyte adhesion, vascular smooth muscle cell proliferation, and fibroblast activation.[19] These activated endothelial cells transdifferentiate into mesenchymal cells with prominent functional abnormalities, such as impaired responsiveness to vasodilators, including nitric oxide and prostacyclins. In addition, activated platelets release thromboxane-A2, platelet-derived growth factor, and transforming growth factor-β (TGF-β), as well as activate thrombin, causing coagulation, thrombosis, vasoconstriction, and fibroblast activation. The initial vascular insult ultimately results in tissue hypoxia. Patients with systemic sclerosis exhibit impaired compensatory vascular repair mechanisms, widespread microangiopathy, microvasculature loss, and diminished angiogenesis, contributing to chronic tissue hypoxia and oxidative stress.
Immune dysregulation and inflammation play a crucial role in the pathogenesis of systemic sclerosis, with dysregulation of both innate and humoral immune systems. Activated T cells are predominant in the tissue and peripheral blood in systemic sclerosis patients. Patients with systemic sclerosis have an imbalance of types 1 and 2 helper T cells (Th1/Th2) cytokines with a predominant Th2 profile. This Th2 profile skewing contributes to increased fibrosis through enhanced collagen synthesis and myofibroblast transdifferentiation, driven by pro-fibrotic cytokines such as TGF-β, interleukins (ILs) 4, 5, and 13, and less anti-fibrotic cytokines such as interferon-γ.
Activated macrophages, monocytes, and dendritic cells further promote vascular injury and fibrosis by activating T- and B cells and producing pro-fibrotic and pro-inflammatory cytokines. Nearly all patients with systemic sclerosis exhibit humoral autoimmunity and the presence of autoantibodies. Activated B cells are responsible for producing these autoantibodies, which are considered directly pathological and serve as important diagnostic markers for the disease.[20] Furthermore, specific autoantibody profiles are associated with distinct disease phenotypes in systemic sclerosis. Vascular injury and inflammation resulting from autoimmunity contribute to tissue fibrosis through mesenchymal cell activation and differentiation. This process leads to irreversible extracellular matrix accumulation, uncontrolled fibroblast activation, the persistence of myofibroblasts, an increase in the microvascular pericyte compartment, and pathologic epithelial-mesenchymal transition.
Histopathology
The hallmark of systemic sclerosis is noninflammatory proliferative or obliterative vasculopathy followed by interstitial or vascular fibrosis. Perivascular inflammatory infiltrates of CD4+ T lymphocytes may be observed early in the disease but are absent in long-standing systemic sclerosis cases. The vasculopathy in systemic sclerosis is characterized by bland intimal proliferation, thickening of the basement membrane, capillary rarefaction, loss of vascular endothelial cadherin, platelet aggregation, and microthrombi formation. Vasculitis or immune complex deposition is rare. However, in the later stages of systemic sclerosis, extensive perivascular fibrosis, progressive luminal occlusion, and tissue fibrosis become evident.
In early systemic sclerosis, dermal edema in the skin is prominent, and perivascular inflammatory infiltrates mainly consist of T lymphocytes and monocytes. Dermal fibrosis becomes the predominant feature as the disease progresses to later stages. Histopathological examination typically shows a loss of dermal capillaries and skin appendages such as hair follicles, sweat glands, and sebaceous glands. A marked dermal expansion is characterized by dense collagen and hyaluronic acid accumulation. Additionally, a loss of dermal lymphatics and the subcutaneous adipose layer is commonly observed.
Pulmonary involvement is widespread in systemic sclerosis, mirroring the skin's pathology. Early in the disease, pulmonary involvement manifests with inflammatory changes, leading to fibrosis and vascular damage.[21] Early inflammatory changes affect the alveolar walls, which are characterized by infiltration with lymphocytes, macrophages, and plasma cells. The most common pulmonary involvement in systemic sclerosis is nonspecific interstitial pneumonitis, which exhibits a fairly uniform distribution of fibrosis along with interstitial inflammation and type II pneumocyte hyperplasia. Another pattern, usual interstitial pneumonia, is characterized by a patchy distribution of fibrosis with scattered fibroblastic foci and carries a poorer prognosis. Pulmonary arterial hypertension (PAH) develops due to vasculopathy and the loss of pulmonary microvasculature secondary to progressive pulmonary fibrosis.
Renal involvement in systemic sclerosis is relatively less common. Chronic ischemic changes are frequently observed, although glomerulonephritis is uncommon unless there is an overlapping syndrome. Scleroderma renal crisis, a rare yet life-threatening manifestation of systemic sclerosis, is associated with changes resembling those seen in thrombotic microangiopathy or malignant hypertension.[22] Histologically, onion skinning (intimal proliferation and reduplication of the elastic lamina with luminal narrowing) of the small interlobular and arcuate renal arteries is a characteristic finding in such cases.
Fibrosis is the predominant pathological feature in systemic sclerosis, affecting various organs beyond the skin. Fibrosis can occur in organs such as the gastrointestinal tract (from the mouth to the rectum), thyroid gland, salivary glands, and penile blood vessels. Inflammatory myositis may also manifest in systemic sclerosis alone or overlap syndromes. Cardiac involvement can lead to constrictive pericarditis, pericardial fibrosis or effusions, and patchy myocardial fibrosis as significant findings.
History and Physical
Systemic sclerosis is a complex disorder affecting multiple systems, and its clinical presentation varies widely among individuals. Diffuse cutaneous systemic sclerosis typically presents more severe symptoms and higher mortality rates compared to limited cutaneous systemic sclerosis, often involving internal organs more extensively and severely. Additionally, systemic sclerosis tends to manifest more severely in males, African Americans, and individuals with a later age of onset.
Raynaud Phenomenon
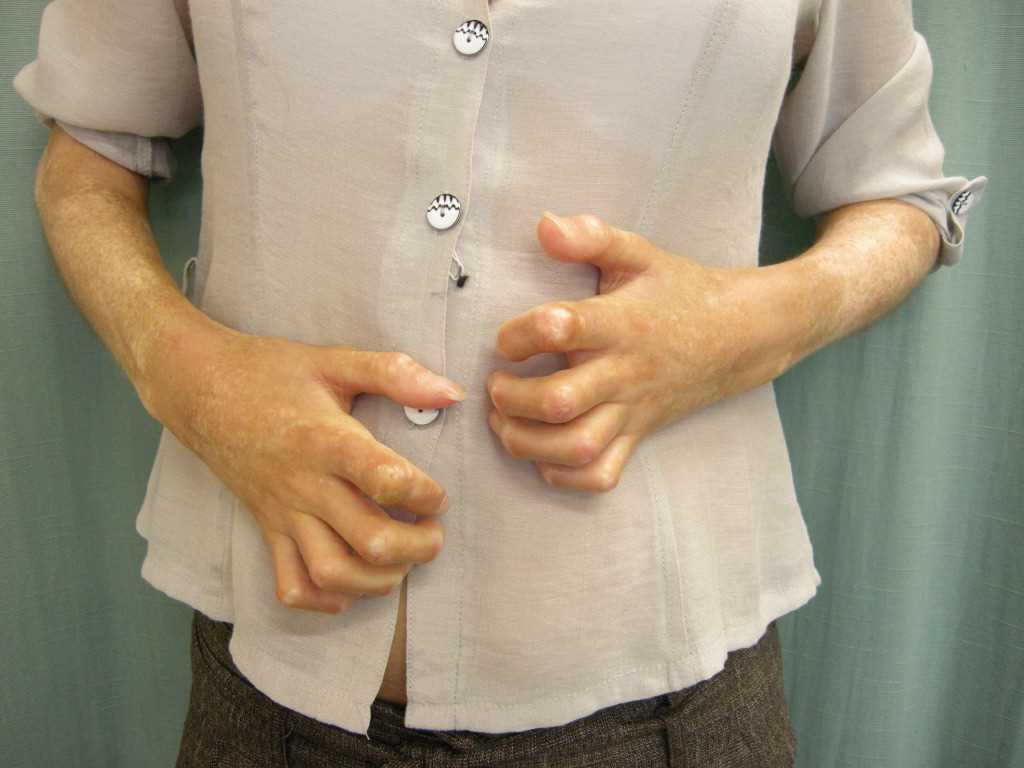
Raynaud phenomenon is an early and common manifestation observed in over 95% of patients with systemic sclerosis.[23] The condition involves vasospasms triggered by exposure to cold, resulting in triphasic color changes, typically affecting the digits, especially in the upper extremities. These color changes may also manifest in areas such as the ears, nose, or tongue. The sequence of color changes includes initial pallor with well-defined white discoloration, followed by ischemic changes with a dusky bluish appearance, and finally, reactive hyperemia leading to red discoloration (see Image. Bilateral Signs of Raynaud Phenomenon in a Patient's Hands). Notably, not all patients report all 3 color phases. Raynaud phenomenon can manifest years before the onset of visceral involvement, particularly in cases of limited cutaneous systemic sclerosis. However, in diffuse cutaneous systemic sclerosis, Raynaud phenomenon may occur simultaneously or shortly after the onset of skin changes. Notably, up to 15% of individuals in the general population experience the Raynaud phenomenon without underlying systemic sclerosis or connective tissue diseases. This is termed primary Raynaud, which is associated with a female predominance (4:1 ratio), early onset (usually occurring in individuals aged 20 or younger), symmetrical mild clinical features, normal nailfold capillaries, negative antinuclear antibodies, and a benign course without ischemic complications. However, secondary Raynaud, as observed in systemic sclerosis, can lead to severe complications.[24]
In systemic sclerosis, the vasospastic insult caused by the Raynaud phenomenon is complicated by the underlying non-vasculitic vasculopathy characterized by intimal hyperplasia and fibrosis. Additionally, abnormal regulation of local vasomotor control contributes to the loss of vessel flexibility. Consequently, digital manifestations such as pitting, ulcers, and tissue loss frequently occur in systemic sclerosis, often leading to digital ischemia and dry gangrene, which can necessitate autoamputation. Moreover, secondary infections are common complications. In systemic sclerosis, Raynaud phenomenon is further characterized by an abnormal nailfold capillary examination, revealing features such as capillary dilation, hemorrhages, and dropout.
Cutaneous Manifestations
Skin involvement is the most overt feature of systemic sclerosis and is present in almost all patients with systemic sclerosis, varying in degree and severity.[25] The classification of systemic sclerosis into limited and diffuse cutaneous subtypes is determined by the degree of cutaneous involvement. Limited cutaneous systemic sclerosis is characterized by skin involvement distal to the elbows and knees, with the trunk typically spared. In contrast, diffuse cutaneous systemic sclerosis involves skin proximal to the elbows, knees, and/or the trunk. Facial involvement may occur in both limited and diffuse cutaneous systemic sclerosis presentations.
During the initial "puffy finger phase," inflammation and non-pitting edema of the hands are prevalent and may persist for several months. Symptoms such as pruritus, burning pain, and erythema are frequently reported. The edema can result in compression of underlying structures, leading to common compression neuropathies such as carpal tunnel syndrome are common. Loss of skin appendages contributes to dry and uncomfortable skin. Subsequently, skin thickening and fibrosis develop during this initial phase, followed by the fibrotic phase.
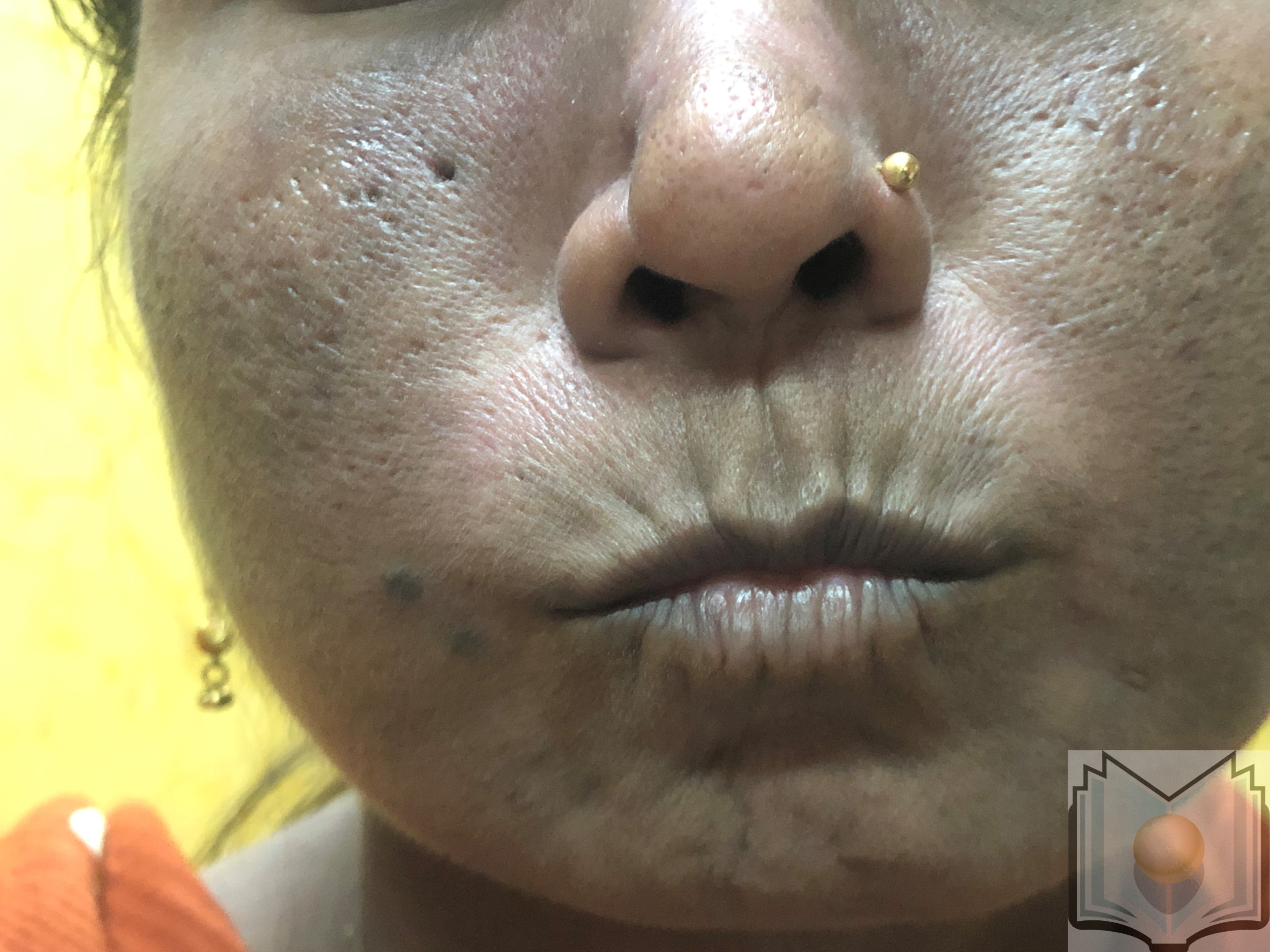
During this second prolonged fibrotic phase, skin fibrosis and thickening commence distal to the metacarpophalangeal joints, a condition termed sclerodactyly, and progressively advance proximally. The thick, leather-like skin and fibrosis of deeper subcutaneous structures contribute to permanent contractures and reduced mobility of the peripheral joints. Further loss of skin appendages and subcutaneous adipose tissue (lipodystrophy) may occur. Facial involvement manifests as a small oral aperture, known as fish-mouth or masked facies (see Image. Pursed Mouth in a Female Patient With Scleroderma).
Skin ulcers may develop at sites of trauma, such as the extensor surfaces of the metacarpophalangeal, interphalangeal, or elbow joints. Additionally, a salt-and-pepper-like appearance, characterized by areas of depigmentation among normally pigmented skin, may be observed. The final skin-softening phase may occasionally occur many years after the initial presentation. During this phase, the skin, particularly on the trunk and upper arms, may soften and return to a clinically normal appearance, although the underlying subcutaneous tissue remains fibrotic.
Telangiectasias, attributed to capillary dilatation, are a common feature of systemic sclerosis and are often observed on the hands, face, mucosal surfaces, and occasionally on the trunk. These telangiectasias blanch upon pressure application and may progressively increase over time. Their presence is associated with a heightened risk of PAH. Subcutaneous calcinosis, although prevalent in both diffuse and limited cutaneous systemic sclerosis, is more frequently encountered in the latter and among patients testing positive for anti-centromere antibodies.[26] Calcinosis arises from localized deposits of calcium hydroxyapatite in the subcutaneous tissue and typically manifests over areas prone to trauma, such as finger pads and the extensor surfaces of elbows. This calcinosis predisposes to skin ulceration and secondary infection.
Musculoskeletal Manifestations
Musculoskeletal manifestations are prevalent in nearly all systemic sclerosis patients. Among these, arthralgia and myalgia are frequently reported. Inflammatory arthritis with true synovitis may occur, often resembling the pattern observed in rheumatoid arthritis, affecting joints such as the metacarpophalangeal, proximal interphalangeal, wrist, and ankle joints. Although erosions are uncommon, they may occur, particularly in association with periarticular calcinosis. While large joint involvement is rare, it can manifest in diffuse cutaneous systemic sclerosis cases. Additionally, there is evidence of systemic sclerosis overlapping with rheumatoid arthritis in up to 5% of cases.[27][28]
Distal bone resorption and osteolysis are seen in the late stages of diffuse cutaneous systemic sclerosis. While hand joint contractures are common, large joint contractures are rare in this form of the disease. Tendon friction rubs, resulting from inflammation, edema, and fibrosis of the tendon sheath, may occur in up to 30% of cases, particularly in diffuse cutaneous systemic sclerosis, and are associated with an unfavorable prognosis. Muscle involvement or myopathy in systemic sclerosis is multifactorial. Approximately 10% of patients exhibit an inflammatory myopathy akin to polymyositis, characterized by rapidly progressive proximal muscle weakness, elevated muscle enzymes, and biopsy findings consistent with inflammatory myositis. Additionally, decreased muscle strength can arise from malnutrition or deconditioning due to joint disease and skin fibrosis.
In systemic sclerosis, direct muscle involvement can result in a fibrosing myopathy characterized by muscle fibrosis and atrophy. Patients with this condition may exhibit mildly elevated muscle enzyme levels, and muscle biopsy typically reveals fibrosis and atrophy with minimal or no inflammation. Unlike inflammatory myopathies, these patients usually do not respond to anti-inflammatory agents. Overall, myopathy in systemic sclerosis is associated with a poor prognosis.
Gastrointestinal Manifestations
Gastrointestinal involvement is almost universal in systemic sclerosis, encompassing diffuse and limited cutaneous systemic sclerosis. Symptoms can range from mild to severe, and any segment of the gastrointestinal tract may be affected. Oropharyngeal involvement manifests as perioral skin tightening, reduced oral aperture, periodontitis, and gingivitis. Dry mouth often arises from salivary gland fibrosis secondary to systemic sclerosis or inflammatory infiltrates associated with secondary Sjögren syndrome. Esophageal dysmotility, primarily due to fibrosis affecting the distal two-thirds of the esophagus, frequently results in dysphagia and heartburn, affecting up to 90% of systemic sclerosis patients.
The lower esophageal sphincter becomes hypotonic, exacerbating reflux symptoms. Complications of esophageal dysmotility may include esophagitis, esophageal strictures, Barrett's esophagus, and bleeding. Gastric involvement may manifest as delayed gastric emptying (gastroparesis), resulting in early satiety, bloating, nausea, vomiting, and anorexia, potentially leading to malnutrition and weight loss.[29][30]
Gastric antral vascular ectasia, also known as "watermelon stomach," results from gastric telangiectasias and can lead to occult or large amounts of gastrointestinal bleeding. Intestinal dysmotility can cause diarrhea, constipation, and intestinal bacterial overgrowth. Mucosal telangiectasias can result in occult gastrointestinal bleeding. Intestinal dysmotility can lead to serious pseudo-obstruction. Wide-mouthed diverticula due to intestinal mucosal muscular atrophy are unique to systemic sclerosis. Reduced anal sphincter tone can result in rectal prolapse and incontinence. Hepatic involvement is not typically associated with systemic sclerosis, although primary biliary cirrhosis has been reported in some cases.[31]
Pulmonary Manifestations
Pulmonary disease is the leading cause of mortality in patients with systemic sclerosis. The characteristic pulmonary involvement includes interstitial lung disease and/or PAH. Rarer pulmonary manifestations encompass pleuritis, obstructive airway disease, aspiration pneumonia, pulmonary hemorrhage, and cryptogenic organizing pneumonia. The spectrum of pulmonary disease in systemic sclerosis spans from clinically asymptomatic conditions to progressive respiratory failure, entailing severe morbidity.[32]
Interstitial lung disease manifests as bibasilar pulmonary fibrosis and is notably more prevalent and severe in diffuse cutaneous systemic sclerosis, African Americans, males, and individuals with anti–topoisomerase I antibodies. Clinically significant interstitial lung disease is present in about 50% of systemic sclerosis cases, although the condition may remain asymptomatic. Postmortem examinations have revealed interstitial lung disease in approximately 80% of systemic sclerosis patients. Interstitial lung disease usually manifests within the initial 4 to 5 years after a systemic sclerosis diagnosis. While nonspecific interstitial pneumonitis is more prevalent than usual interstitial pneumonia, a mixed pattern is also observable. Symptoms include dyspnea, fatigue, and, later in the disease course, a non-productive cough.
Pulmonary function test reveals a restrictive pattern with decreased lung volumes and FEV1/FVC ratio. Diffusing capacity for carbon monoxide (DLCO) may be reduced, partly due to the underlying pulmonary vascular disease. High-resolution computed tomography (HRCT) of the chest is highly sensitive, particularly in the early stages of the disease, revealing increased subpleural lung attenuations in the bilateral posterior basal areas. Ground-glass opacities may indicate alveolitis, while additional findings include honeycombing, traction bronchiectasis, and thickening of the interlobular septa.[33]
PAH represents another common pulmonary manifestation, with a spectrum ranging from asymptomatic disease to severe PAH accompanied by right heart failure. Typically, PAH manifests late in the course of the disease, often occurring more than 10 years after the initial diagnosis of systemic sclerosis. This condition can affect 30% to 50% of individuals with systemic sclerosis, with a higher incidence observed in cases of limited cutaneous systemic sclerosis. Risk factors for PAH in systemic sclerosis include a later age at diagnosis, multiple telangiectasias, and the presence of anti–U3-RNP antibodies. Symptoms may include dyspnea initially, followed by chest pain, lower extremity swelling, lightheadedness, and syncope as the disease progresses. An accentuated "P component" of the second heart sound (S2) may be detectable.
A pulmonary function test reveals an isolated reduction in diffusion capacity. Additionally, levels of NT-proBNP in the blood may be elevated. On echocardiography, an elevated peak right ventricular systolic pressure may be observed. When PAH is suspected, a right heart catheterization should be performed to confirm the diagnosis and exclude other potential causes. A diagnosis of PAH is typically confirmed by an elevation in mean pulmonary arterial pressure to 25 mm Hg or higher, along with a normal pulmonary capillary wedge pressure.
Cardiac Manifestations
Cardiac manifestations include pericarditis, pericardial effusion, dilated cardiomyopathy, and arrhythmias. Left ventricular diastolic dysfunction can occur secondary to PAH. Cardiac involvement is associated with an increased risk of infection and a poor prognosis.[34] Pericardial effusions are usually small and exudative, and tamponade is uncommon. Pericardial effusions are more common in diffuse cutaneous systemic sclerosis and are predictive of scleroderma renal crisis. Dilated cardiomyopathy arises from patchy myocardial fibrosis. Fibrosis within the conduction pathways may precipitate arrhythmias, with premature ventricular contractions being the most common, although heart block, intraventricular conduction delays, and supraventricular tachycardia have also been reported.
Renal Manifestations
Renal manifestations in systemic sclerosis are exemplified by scleroderma renal crisis, historically almost always fatal and the primary cause of mortality in systemic sclerosis before the discovery of angiotensin-converting enzyme (ACE) inhibitors.[22] Scleroderma renal crisis occurs in approximately 10% of systemic sclerosis patients, predominantly in those with diffuse cutaneous systemic sclerosis, and typically manifests within three years of diagnosis. Risk factors for the development of scleroderma renal crisis include African-American race, pericardial effusion, tendon friction rubs, new-onset anemia, diffuse cutaneous systemic sclerosis, anti-RNA polymerase III antibody, and high-dose (or chronic low-dose) corticosteroids. Indicators of poor outcomes in scleroderma renal crisis encompass male gender, advanced age at onset, and serum creatinine levels exceeding 3 mg/dL.[35]
The pathology of scleroderma renal crisis primarily involves vasculopathy, akin to the vascular changes observed in other organs affected by systemic sclerosis rather than glomerulonephritis. Clinically, it manifests with new-onset hypertension or malignant hypertension, often accompanied by renal insufficiency. Microangiopathy associated with this condition frequently results in hemolytic anemia and thrombocytopenia. While concurrent proteinuria may occur, microscopic hematuria is often observed but tends to be mild.
Other Manifestations
Other manifestations of systemic sclerosis include hypothyroidism, which occurs in up to 15% of patients, particularly those with limited cutaneous systemic sclerosis, often due to thyroid gland fibrosis. Moreover, autoimmune thyroid diseases, such as Hashimoto thyroiditis and Graves disease, are more prevalent among systemic sclerosis patients. These individuals also have a higher risk of developing other autoimmune conditions, such as primary biliary cirrhosis and secondary Sjögren syndrome. Additionally, thrombocytopenia has been observed in some cases.[36]
Overlap syndromes are common in systemic sclerosis and can involve conditions such as rheumatoid arthritis and polymyositis. Additionally, patients with systemic sclerosis have an elevated risk of psychological disorders such as depression, affecting up to 50% of individuals. Another variant, systemic sclerosis sine scleroderma, is characterized by internal organ manifestations of systemic sclerosis, particularly in the pulmonary and gastrointestinal systems, without skin thickening. These patients typically exhibit the Raynaud phenomenon, along with abnormal nailfold capillary exam results and positive antinuclear antibodies.
Classification Criteria
In 2013, the American College of Rheumatology published the updated classification criteria for systemic sclerosis, offering improved sensitivity and specificity compared to the older 1980 criteria.[37] Although primarily designed for clinical study inclusion, these criteria can be applied cautiously in clinical settings. Utilizing a scoring system, a score of 9 or higher indicates classification as systemic sclerosis.
- Bilateral skin thickening proximal to metacarpophalangeal joints: 9
- Skin thickening of fingers (counting only higher score)
- Between distal and proximal interphalangeal joints: 4
- Puffy fingers: 2
- Fingertip lesions (counting only the higher score)
- Fingertip pitting scars: 3
- Digital tip ulcers: 2
- Telangiectasia: 2
- Abnormal nailfold capillaries: 2
- Raynaud phenomenon: 3
- Lung disease (maximum score of 2)
- Positive systemic sclerosis-specific antibodies (anti-centromere, anti–Scl–70, and anti–RNA polymerase III): 3
Evaluation
Systemic sclerosis is diagnosed through clinical assessment. Early detection of the disease, evaluation of the extent of involvement, and ongoing surveillance for internal organ manifestations are essential for its effective management.
Clinical Evaluation
Assessment of skin thickness is performed using the modified Rodnan skin score, which assigns scores ranging from 0 to 3, with 0 indicating uninvolved areas and 3 indicating severe skin thickening. Monitoring changes in the score over time is important as it holds prognostic significance. Nailfold capillary examination is warranted in all patients presenting with Raynaud phenomenon and suspected systemic sclerosis. A comprehensive physical examination of patients targeting multiple organ systems is conducted during each visit to detect underlying organ involvement. Regular blood pressure monitoring, both in the clinic and at home, is highly recommended, particularly in individuals recently diagnosed with diffuse cutaneous systemic sclerosis, those experiencing new-onset hypertension, or those with a significant worsening of existing hypertension, as it may signal the onset of scleroderma renal crisis.
Autoantibody Tests
Autoantibodies are an essential diagnostic tool, offering predictive insights into disease phenotype and prognosis. Antinuclear antibodies, detected through direct immunofluorescence, are positive in over 90% of systemic sclerosis cases.[38] In instances where antinuclear antibodies are negative, it is imperative to rule out other potential diagnoses before confirming systemic sclerosis. Specific autoantibodies, with a positivity rate of 60% to 70%, offer further diagnostic specificity. These antibodies, as mentioned below, which are mutually exclusive, can manifest several months to years before the clinical onset of systemic sclerosis.
Anti-centromere antibody: Anti-centromere antibodies target 4 centromere antigens (CENP -B, -A, -C, D) and are predominantly observed in limited cutaneous systemic sclerosis, although they may occasionally occur in diffuse cutaneous systemic sclerosis. They can also manifest in conditions such as Sjögren syndrome and systemic lupus erythematosus. Anti-centromere antibodies are associated with an increased risk of PAH. Furthermore, they are linked to limited cutaneous involvement, a reduced likelihood of interstitial lung disease, and generally better survival rates compared to other autoantibodies.
Anti-topoisomerase I (Scl-70) antibody: Anti-topoisomerase I (Scl-70) antibodies target the catalytic region of DNA helicase topoisomerase I. They are predominantly observed in diffuse cutaneous systemic sclerosis and infrequently in limited cutaneous systemic sclerosis. The presence of anti–Scl-70 antibodies is linked to an elevated risk of diffuse cutaneous involvement, interstitial lung disease, and cardiac involvement.[39]
Anti-RNA polymerase III antibody: Anti-RNA polymerase III antibodies target the eukaryotic RNA polymerase III. They are particularly associated with diffuse cutaneous systemic sclerosis and are linked to rapidly progressing and aggressive diffuse skin involvement, poor cutaneous outcomes, and scleroderma renal crisis. Additionally, these antibodies are also associated with a lower risk of interstitial lung disease and PAH. Some studies have indicated an association between anti-RNA polymerase III antibodies and malignancies in systemic sclerosis patients.
Anti–U3-RNP (fibrillarin) antibody: Anti-U3-RNP (fibrillarin) antibodies are prevalent in African Americans and are associated with an overall poor prognosis in systemic sclerosis. Their presence correlates with increased internal organ involvement, diffuse cutaneous manifestations, interstitial lung disease, PAH, scleroderma renal crisis, myositis/myopathy, and cardiac complications.[40]
Other autoantibodies: Other autoantibodies include anti-Th/To antibodies that are associated with limited skin disease. Anti–PM/Scl antibodies associated with limited skin disease and overlap syndrome predispose to inflammatory myositis and interstitial lung disease. Anti–U1-RNP antibodies are more prevalent in African Americans and are associated with overlap syndrome and mixed connective tissue disease. This antibody profile is linked to limited cutaneous involvement and increased risk of inflammatory arthritis, myositis, lupus skin rashes, and lupus nephritis. Anti-Ku antibodies are also associated with overlap syndrome in systemic sclerosis, which is associated with more inflammatory arthritis and myositis.
Laboratory Evaluation
Complete blood counts to assess for anemia may be multifactorial.[41] Renal function and 24-hour urine protein or urine protein:creatinine ratio should be closely monitored. Although inflammatory markers typically do not provide significant diagnostic significance, notable elevations may indicate active inflammatory myopathy or inflammatory arthritis. Muscle enzymes, including creatine kinase and aldolase, may be elevated, especially in the presence of inflammatory myopathy.
Ancillary and Radiographic Evaluation
X-rays of the extremities can reveal calcinosis and loss of distal phalanges. Periarticular erosions are rare and uncommon, although periarticular osteopenia can be seen. Musculoskeletal ultrasound can be used to show features of tenosynovitis. When muscle involvement is suspected, electromyography/nerve conduction velocity testing is the initial diagnostic choice. If abnormal results are obtained, a muscle biopsy may be warranted. High-resolution CT (HRCT) scan is the preferred imaging modality for assessing interstitial lung disease, as it can detect subtle findings that may not be visible on a chest x-ray.[29]
Pulmonary function tests, including spirometry, lung volumes, and diffusion capacity, can detect a restrictive pattern in interstitial lung disease very early in the disease process. A lung biopsy is of very limited value and should be avoided unless other diagnostic concerns are concurrent. When PAH is suspected, transthoracic echocardiography is usually initially performed, although right heart catheterization should follow to confirm the diagnosis and rule out other etiologies. Electrocardiography and Holter monitoring can be useful in detecting arrhythmias.
Pericardial effusions can be well visualized on transthoracic echocardiography. In patients suspected of myocardial involvement, a cardiac MRI may be necessary. When esophageal and upper gastrointestinal involvement is suspected, upper gastrointestinal endoscopy, esophageal manometry, barium swallow studies, and a 24-hour pH probe can be utilized.[42] A characteristic finding indicative of esophageal dysmotility is a dilated esophagus with excessive air observed on a CT scan.
Treatment / Management
No definitive treatment or universally accepted disease-modifying agent can alter the natural course of the disease. However, managing the affected system or systems has proven effective. Early diagnosis is crucial for achieving improved outcomes. Clinical evaluation and identification of affected organs and disease progression are critical for treatment efficacy. Furthermore, treatment goals must be holistic and tailored to optimize the quality of life for affected patients while also preventing further organ damage. Patient education about the disease and encouragement to engage in regular exercise, maintain a healthy diet and lifestyle, and seek emotional support should be considered for every individual with systemic sclerosis.
Several agents have been investigated for different manifestations of systemic sclerosis, yet a generalized lack of large randomized controlled trials undermines their efficacy. Cyclophosphamide (for lung disease and skin disease), mycophenolate mofetil (for lung disease and skin disease), methotrexate (MTX; for skin disease, inflammatory arthritis, and myositis), azathioprine (for skin disease, lung disease, and myositis), and hydroxychloroquine (HCQ; for skin disease) stand out as some of the most frequently utilized immunosuppressive agents.
Cyclosporine (for skin disease), infliximab (for inflammatory arthritis), and rituximab (for skin disease and lung disease) have limited available data. Corticosteroids should generally be avoided in systemic sclerosis due to the risk of precipitating scleroderma renal crisis. High-dose corticosteroids and long-term use of low to moderate-dose corticosteroids have been associated with the precipitation of scleroderma renal crisis. Therefore, these medications should only be considered in cases of absolute necessity, such as refractory inflammatory myositis, inflammatory arthritis, or active inflammatory alveolitis, and then used at the lowest possible dose for the shortest duration possible.
Specific Therapies in Scleroderma
Raynaud phenomenon: Raynaud phenomenon treatment aims to prevent digital ischemia and ulcers, which are easier to prevent than treat. Conservative management for the Raynaud phenomenon is still the cornerstone of management, and patients are advised to keep their extremities and body warm, avoid smoking and stress, and eliminate sympathomimetic medications when possible. In cases where β-blockers are used, switching to an alternative medication should be attempted if possible, as β-blockers may exacerbate Raynaud symptoms.
Vasodilator therapy has demonstrated efficacy in Raynaud syndrome. Dihydropyridine calcium channel blockers, such as nifedipine (30-120 mg/d) or amlodipine (5-20 mg/d), are the first-line agents. In addition, vasodilators, including pentoxifylline, nitroglycerin, and phosphodiesterase inhibitors such as sildenafil (20 mg once daily to 3 times daily) may be considered. Prostacyclin analogs (iloprost) and endothelin receptor antagonists (bosentan) can be utilized for refractory cases with digital ischemic ulcers. Typically, iloprost is administered for 3 to 5 consecutive days at a continuous dose of 0.5 to 2 ng/kg/min, titrated to the maximum tolerated dose.[43]
The infusion can be continuous in the inpatient setting or administered over 8 hours in the outpatient setting. Bosentan is typically initiated at 62.5 mg twice daily for 4 weeks and may be increased to 125 mg twice daily. In refractory cases, proximal or distal sympathectomy may be considered. While botulinum toxin injections have shown efficacy in some small studies, their overall effectiveness remains limited. Vasodilator pumps may be utilized for patients unable to tolerate vasodilator therapies. Effective wound care is essential in the management of the condition in patients with digital ulcers.
Skin disease: Several immunosuppressive agents have been used in sclerodactyly, including MTX, HCQ, mycophenolate mofetil, and cyclophosphamide. MTX is usually administered orally or subcutaneously at a dose of 15 to 25 mg per week. When systemic glucocorticoids are prescribed, they are usually initiated concurrently with MTX. The recommended regimen consists of either intravenous (IV) methylprednisolone at a dosage of 30 mg/kg/d for 3 consecutive days per month or oral prednisone at a dosage of 1 mg/kg/d. Patients are often treated with MTX for a duration of 1 to 2 years. Following 6 to 12 months of documented inactivity, gradual tapering of the MTX dose (reduction by 2.5 mg every 2 to 4 weeks) is typically initiated. However, relapses may occur following cessation of therapy.[44]
IV methylprednisolone is typically discontinued after 3 to 4 months of symptom resolution. Oral prednisone is tapered slowly over 3 to 4 months. While lacking robust data, most rheumatologists prefer mycophenolate mofetil for moderate-to-severe skin thickening. The typical dosage is 1 g twice daily, with a maximum of 1.5 g twice daily. Treatment is continued for 6 to 12 months after symptom resolution, tapered over several months. Cyclophosphamide is recommended for patients refractory to MTX (15-25 mg weekly) or mycophenolate mofetil (1.5-3 g daily) or with progressive disease.[45]
Antihistamines and topical moisturizing agents can help with pruritus. Topical therapy with corticosteroids, vitamin D analogs, and tacrolimus can also be used for limited skin disease.[46] Telangiectasias usually do not require any treatment, although laser therapy can be considered for cosmetic purposes. No medical treatment has been proven effective in calcinosis, and surgical debulking may provide some relief in severe cases.
Musculoskeletal involvement: Mild arthralgias in most patients often do not require treatment or respond well to nonsteroidal anti-inflammatory drugs (NSAIDs). Low-dose glucocorticoids (<10 mg/d of prednisone or equivalent) may be used for less than 2 to 4 weeks initially to control arthritis. Inflammatory arthritis can be managed with disease-modifying antirheumatic drugs such as HCQ (200-400 mg daily) and MTX. MTX is added if patients do not respond to HCQ.[47] Antitumor necrosis factor agents can be used in refractory severe inflammatory arthritis.[48]
Physical and occupational therapy are crucial in preventing contractures. Management of inflammatory myositis in systemic sclerosis is similar to polymyositis, but high-dose corticosteroids should be avoided. Immunosuppressive agents, such as MTX and azathioprine, are effective in managing inflammatory myositis. Treating noninflammatory myopathy in systemic sclerosis is challenging, and physical therapy and exercises are more beneficial than immunosuppressive agents.
Pulmonary involvement: Pulmonary involvement in systemic sclerosis is now the leading cause of mortality, necessitating interprofessional involvement from rheumatologists, pulmonologists, and cardiologists. Early detection is crucial for managing interstitial lung disease in systemic sclerosis. While cyclophosphamide has shown benefit for up to 18 months, its efficacy diminishes by 24 months. Some small studies have demonstrated the efficacy of cyclophosphamide followed by azathioprine for maintenance therapy.
Mycophenolate mofetil (with a target dose of 1.5-3 g daily in 2 divided doses) has shown benefits in managing systemic sclerosis-related interstitial lung disease. Lung transplantation may be necessary for carefully selected individuals. Nintedanib, a tyrosine kinase inhibitor, was approved by the US Food and Drug Administration (FDA) for managing interstitial lung disease in systemic sclerosis in 2019, as clinical trials showed a slowing of the decline of pulmonary function tests in patients with systemic sclerosis with interstitial lung disease. Other investigational modalities include stem cell transplants and antifibrotic agents.
When no contraindication exists, supplemental oxygen, diuretics, and anticoagulation are recommended, alongside patient education, a healthy lifestyle, and exercise as tolerated. Calcium channel blockers are generally ineffective. Nifedipine (starting dose 30 mg/d), amlodipine (starting dose 2.5 mg/d), and extended-release diltiazem (starting dose 120 mg/d) can be titrated up to the maximum tolerated dosages over weeks.
Vasodilator therapy is recommended and may include phosphodiesterase-5 inhibitors (such as tadalafil 40 mg daily and sildenafil 20 mg orally 3 times daily), endothelin receptor antagonists (such as bosentan at 62.5-125 mg twice daily or sitaxsentan, ambrisentan, or macitentan), and/or prostacyclin analogs (such as epoprostenol, treprostinil, beraprost, and iloprost at 2.5-5 μg inhaled 6-9 times daily). Prostacyclin therapy is considered the most effective, although all these agents improve hemodynamics and quality of life. Combination therapy may be considered in patients who fail to improve on monotherapy or have severe PAH.
Cardiac involvement: Arrhythmias are managed with antiarrhythmic agents and, occasionally, pacemaker placement. Currently, evidence supporting the efficacy of immunosuppressive agents or vasodilator therapy does not exist in scleroderma-related cardiac involvement.
Gastrointestinal involvement: Exercise may aid in improving the oral aperture while maintaining good dental hygiene, which is essential to prevent caries and cavities due to sicca symptoms. Sugar-free lozenges and secretagogues such as pilocarpine or cevimeline can also alleviate dry mouth. For patients experiencing heartburn and gastroesophageal reflux, lifestyle and dietary adjustments, such as elevating the head of the bed, avoiding late and large meals, steering clear of spicy foods, and opting for small, frequent meals, are recommended. NSAIDs should be avoided. Proton pump inhibitors are preferred over H2 blockers, with high doses permissible, particularly for patients with erosive esophagitis. A combination of proton pump inhibitors and H2 blockers may be considered in severe cases.[30]
Motility agents such as metoclopramide and proton pump inhibitors may benefit patients with gastroparesis. Laser coagulation is a viable option for managing bleeding due to gastric antral vascular ectasia. Patients diagnosed with small intestinal bacterial overgrowth (SIBO) syndrome may require rotational antibiotics for effective management.
Scleroderma renal crisis: ACE inhibitors represent the sole effective treatment for scleroderma renal crisis. Initiation should occur upon the earliest signs, with a maximum tolerated dose utilized. While any ACE inhibitor is viable, captopril is preferred due to its dosing flexibility. Currently, no data support the efficacy of angiotensin receptor blockers or renin inhibitors in scleroderma renal crisis.[49]
Renal function may initially decline, but continuation of the ACE inhibitor is crucial. With appropriate management, renal function can markedly improve, albeit over several months to years, potentially allowing for discontinuation of dialysis. However, prophylactic use of ACE inhibitors is not recommended, as it does not prevent scleroderma renal crisis and is associated with increased morbidity and mortality.
Differential Diagnosis
Systemic sclerosis is primarily a clinical diagnosis. However, several other diseases that can mimic systemic sclerosis should be considered when establishing a differential diagnosis.
Eosinophilic Fasciitis
Eosinophilic fasciitis is characterized by eosinophilic inflammation of the deep fascia, resulting in thickening and a woody induration of the upper and lower extremities, excluding the hands and feet. Raynaud phenomenon is not associated with eosinophilic fasciitis, and nailfold capillary examination typically appears normal. Antinuclear antibodies and specific autoantibodies are generally negative in eosinophilic fasciitis cases. Patients may develop contractures resembling those seen in systemic sclerosis. Eosinophilic fasciitis may be associated with underlying malignancies. Histologically, a skin biopsy often reveals eosinophilic infiltrates in the deep fascia.[50]
Scleromyxedema
Scleromyxedema is usually seen in patients with monoclonal gammopathy or multiple myeloma and is characterized by papular waxy lesions on the face, neck, extremities, and fingers. Patients may also exhibit associated symptoms such as seizures and dementia. Raynaud phenomenon is not a feature of scleromyxedema, and nailfold capillary examination typically appears normal. Antinuclear antibodies and specific autoantibodies are typically negative in scleromyxedema cases. Histologically, a skin biopsy of scleromyxedema often reveals dermal fibrosis accompanied by perivascular inflammation and the deposition of mucin and fibrocytes, which are not typical findings in systemic sclerosis.[51]
Scleredema
Scleredema can be associated with conditions such as diabetes mellitus, monoclonal gammopathy, fatigue, infections, and malignancies. This condition is characterized by the doughy, indurated appearance of the skin on the neck, back, and face, with digits typically being spared. Raynaud phenomenon is not associated with scleredema, and the nailfold capillary examination appears usually normal. Antinuclear antibodies and specific autoantibodies are generally negative in scleredema cases. Histologically, a skin biopsy of scleredema often reveals dermal fibrosis without perivascular inflammation, along with mucin deposition.[52]
Nephrogenic Systemic Fibrosis
Nephrogenic systemic fibrosis is a rare phenomenon observed in patients with end-stage renal disease following exposure to gadolinium contrast. This condition is characterized by cobblestone-like nodular plaques on the extremities, trunk, hands, and feet while sparing the face. Raynaud phenomenon is not associated with nephrogenic systemic fibrosis, and the nailfold capillary examination typically appears normal. Antinuclear antibodies and specific autoantibodies are generally negative in nephrogenic systemic fibrosis cases. Histologically, a skin biopsy of nephrogenic systemic fibrosis often reveals dermal and epidermal fibrosis without perivascular inflammation, accompanied by the deposition of mucin and fibrocytes.
Eosinophilia Myalgia Syndrome
This syndrome was an epidemic associated with the use of L-tryptophan, resulting in severe myalgias, visceral involvement, and elevated CK levels.
Toxic Oil Syndrome
This syndrome was also an epidemic in Spain associated with the consumption of adulterated rapeseed oil, causing livedo reticularis, pulmonary infiltrates, and elevated levels of creatine kinase.
Prognosis
Systemic sclerosis is associated with high mortality, having the highest case-specific mortality among all collagen vascular disorders. Previously, scleroderma renal crisis was the most common cause of mortality before the advent of ACE inhibitors. The use of ACE inhibitors and increased awareness have significantly reduced scleroderma renal crisis-related mortality. Currently, pulmonary disease is the most common cause of mortality in systemic sclerosis patients.[53]
Over the past 30 years, the prognosis of systemic sclerosis has improved, with 5-year survival rates reaching up to 80%. However, patients with advanced PAH have a less than 50% 2-year survival rate. Additionally, patients with systemic sclerosis-related PAH have a poorer prognosis compared to those with idiopathic PAH.
Before the discovery of ACE inhibitors, the 1-year survival rate for patients experiencing scleroderma renal crisis was less than 15%. However, with increased awareness and the use of ACE inhibitors, the 1-year survival rate in scleroderma renal crisis has improved significantly to over 85%. Patients with systemic sclerosis also face a heightened risk of developing malignancies, particularly lung cancer. In addition, an increased risk of esophageal adenocarcinoma exists in systemic sclerosis cases associated with chronic gastroesophageal reflux, which can lead to Barrett esophagus.
Prognostic Factors and Associations in Systemic Sclerosis
- African-American race: Early disease onset, severe disease, interstitial lung disease, and scleroderma renal crisis.
- Late age of diagnosis: PAH and poor outcomes after scleroderma renal crisis.
- Diffuse cutaneous systemic sclerosis: Diffuse skin disease, large joint inflammatory arthritis, tendon friction rubs, joint contractures, distal bone osteolysis, scleroderma renal crisis, interstitial lung disease, and pericardial effusion.
- Limited cutaneous systemic sclerosis: Limited skin disease, calcinosis, PAH, and hypothyroidism.
- Multiple telangiectasias: PAH.
- Pericardial effusion: Scleroderma renal crisis.
- Tendon friction rubs: Scleroderma renal crisis and a poor overall prognosis.
- New-onset anemia: Scleroderma renal crisis.
- Corticosteroid use: Scleroderma renal crisis.
- Anti-centromere antibody: Limited cutaneous systemic sclerosis and PAH. Lower risk of interstitial lung disease.
- Anti–Scl-70 antibody: Diffuse cutaneous systemic sclerosis, interstitial lung disease, and cardiac involvement.
- Anti-RNA polymerase III antibody: Rapidly progressing and aggressive diffuse skin involvement, poor cutaneous outcomes, scleroderma renal crisis, and malignancies. Lower risk of interstitial lung disease and PAH.
Complications
Systemic sclerosis is associated with various complications primarily due to end-organ damage from fibrosis. Digital ischemia may progress to gangrene, necessitating amputation. Gastrointestinal complications can result in malnutrition. Pulmonary involvement is the primary cause of morbidity, causing irreversible pulmonary fibrosis. Patients with scleroderma renal crisis can have permanent renal damage, although the use of ACE inhibitors leads to recovery of renal function in most cases.
Deterrence and Patient Education
Patient education plays a crucial role in disease management, including lifestyle adjustments. Individuals with Raynaud phenomenon should be educated on maintaining body and extremity warmth, avoiding extreme cold exposure, vasoconstrictive agents, and trauma to the digits. Patients with systemic sclerosis should be advised to quit smoking and avoid exposure to secondhand smoke. Regular blood pressure monitoring at home can aid in early detection of scleroderma renal crisis.
Enhancing Healthcare Team Outcomes
Systemic scleroderma results in significant morbidity, causing severe disability without a known cure and only offering symptomatic treatment. Due to its impact on multiple organ systems, a collaborative interprofessional healthcare team approach is necessary. This team should include the patient's primary care clinician, rheumatologists, gastroenterologists, cardiologists, pulmonologists, nephrologists, and dermatologists.
Furthermore, nurses and pharmacists are critical in patient care for systemic sclerosis. Pharmacists ensure that patients are not taking medications causing vasoconstriction and educate them on adhering to blood pressure medications. Patients with scleroderma require close follow-up by healthcare providers. Nursing staff are essential for patient education, monitoring, and follow-up care. They can also coordinate activities among healthcare professionals involved in the patient's treatment. Patient education about managing systemic sclerosis manifestations is vital for preventing long-term morbidity.