Continuing Education Activity
Antiphospholipid syndrome is characterized by the presence of antiphospholipid antibodies in the setting of thrombosis or pregnancy loss or complications. These antibodies are autoantibodies directed against phospholipid-binding proteins and are composed of 3 main categories. Despite in vivo prolongation, these antibodies cause in vivo thrombosis in multiple organ systems. The most common sites of venous and arterial thrombosis are the lower limbs and cerebral arterial circulation, respectively. However, thrombosis can occur in any organ. Catastrophic antiphospholipid syndrome is a dreaded complication with high mortality. Treatment is variable depending on clinical presentation and antibody positivity. This activity reviews the criteria for considering antiphospholipid syndrome in the differential diagnosis and outlines proper evaluation protocols, highlighting the role of the interprofessional team in caring for patients with this condition.
Objectives:
Identify the signs and symptoms of a patient with antiphospholipid syndrome.
Apply best practices in the evaluation of antiphospholipid syndrome.
Implement evidence-based treatment and management strategies for antiphospholipid syndrome depending on the clinical picture and results of laboratory testing.
Collaborate with interprofessional healthcare teams to provide comprehensive care for patients with antiphospholipid syndrome.
Introduction
Antiphospholipid antibodies (APLAs) are autoantibodies that target phospholipid-binding proteins. Antiphospholipid syndrome (APS) is a multisystemic autoimmune disorder.[1] The hallmark of APS comprises the persistent presence of APLAs in the setting of arterial and venous thrombus or pregnancy loss.[2]
The most common sites for venous and arterial thrombosis are the lower limbs and cerebral arterial circulation, respectively. However, thrombosis can occur in any organ. Laboratory tests, including enzyme-linked immunosorbent assay (ELISA) and functional assays, are used to identify APS. The 3 known APLAs include:
- Anticardiolipin antibodies IgG or IgM (ELISA)
- Anti-beta-2-glycoprotein-I (anti-β2GPI) antibodies IgG or IgM (ELISA)
- Lupus anticoagulants (functional clotting assays)
Etiology
APS can be primary when there is no evidence of autoimmune disease or secondary to autoimmune processes, such as systemic lupus erythematosus (SLE), in 40% of cases.[3] A study found positive APLAs in 6% of all pregnant patients, 13.5% of stroke patients, and 9.5% of patients with deep venous thromboses (DVTs).[4]
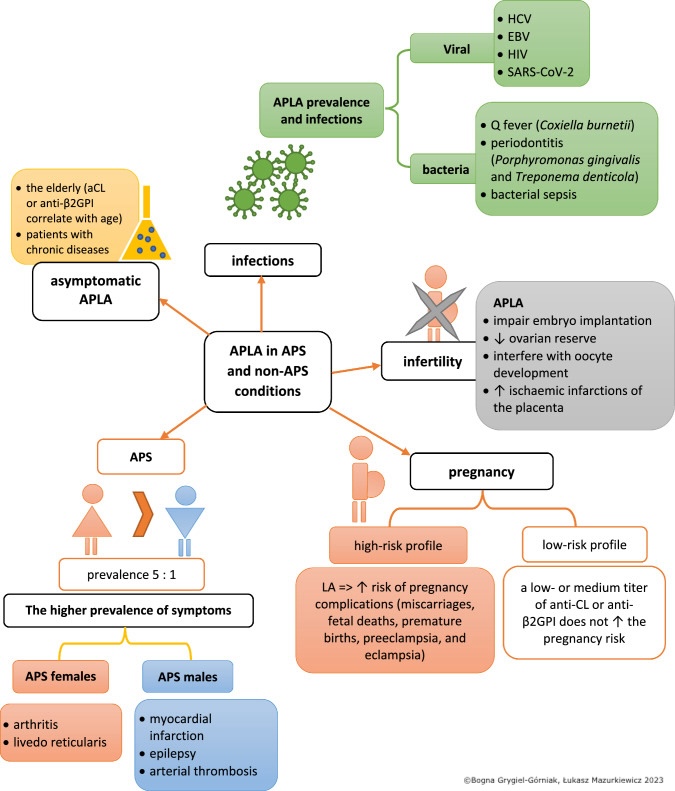
Genetic risk factors, such as coagulation factor mutations, increase the risk of antiphospholipid antibody–associated thrombosis. HLA-DR7, DR4, DRw53, DQw7, and C4 null alleles have been reported to be associated with APS. Infections, particularly viral, are associated with elevated APLA levels compared to bacterial infections. Some of these include Borrelia burgdorferi, Coxiella burnetii, Treponema, hepatitis C, HIV, COVID-19, Epstein-Barr virus (EBV), and Leptospira have been implicated in APLA formation. In fact, a large meta-analysis found that almost 50% of patients diagnosed with COVID-19 had positive APS, most commonly lupus anticoagulant. However, this analysis found no increased thrombotic risk in APLA-positive COVID-19 patients.[4][5]
Several drugs, including chlorpromazine, procainamide, quinidine, and phenytoin, can induce APLA production. Low levels of APLAs may also be normally present and may be transient, leading to the requirement of positive antibodies at least 12 weeks apart for diagnosis.[4]
APS can be further classified based on clinical manifestations, such as obstetric, thrombotic, or both, and whether it involves life-threatening multiorgan involvement.
- Thrombotic APS: Patients are diagnosed with APS based on arterial or venous thrombosis and persistent laboratory criteria for APLA. The most common presentation is a DVT.[6]
- Obstetric APS: Patients are diagnosed based on APS-defining pregnancy morbidity, such as including premature birth due to severe preeclampsia, fetal death after 10 weeks of gestation, placental insufficiency, or multiple embryonic losses before 10 weeks of gestation, and persistent laboratory criteria for APLA. Patients with both thromboembolic complications and APS-defining pregnancy morbidity are recognized as having both thrombotic and obstetric APS.
- Catastrophic APS: This rare and life-threatening form of APS is defined as thrombotic complications affecting multiple organs, both microvascular and macrovascular. See below for further details.[7]
Epidemiology
The incidence of APS is believed to be around 2.1 in 100,000 individuals in the United States, with a prevalence of 50 per 100,000. In Europe, rates are lower, with an incidence of 1.1 per 100,000. Rates are also believed to be lower in Asia; South Korea has an incidence of 0.75 per 100,000, with a prevalence of 6.19 per 100,000.[4]
Low-titer anticardiolipin antibodies can be observed in up to 10% of healthy individuals, and the prevalence of a positive APLA test increases with age. In fact, a study involving centenarians without known autoimmune disease showed that 54% were positive for anti-β2GPI-IgG and 21% were positive for anticardiolipin-IgG. None were positive for the lupus anticoagulant test, possibly making this a more specific marker.[6] However, high titers and persistent positivity are rare among healthy individuals, occurring in less than 1%. Patients with SLE are at a high risk of having a positive APLA test and an APLA-related clinical outcome, such as thrombosis or pregnancy-related morbidity. About 50% to 70% of the patients with SLE with positive APLA progress to APS.[3][8]
APLA positivity has also been demonstrated in up to 20% of patients with rheumatoid arthritis.[9] A study involving 197 couples with a history of frequent abortions found that 20% of them tested positive for APLA.[10] Another study identified the presence of APLA, such as lupus anticoagulant or anticardiolipin antibodies, in 14% of patients with recurrent venous thromboembolism.[11]
Pathophysiology
Although not all patients with APLAs develop APS, a strong association exists between the presence of APLAs and venous thrombosis, myocardial infarction, and ischemic stroke.[12] The antibody profile, including type, titer, and underlying comorbidities, may determine the likelihood of developing clinical APS. Triple positivity with positive lupus anticoagulant and high titers of anticardiolipin and anti-β2GPI antibodies pose a high risk of APS development. In contrast, isolated or intermittent positivity or low titers of anticardiolipin or anti-β2GPI antibodies pose a low risk.[13][14] Patients with SLE, coexisting cardiovascular risk factors, a history of recurrent thrombotic events despite anticoagulation therapy, and a history of arterial thrombosis are also at a high risk of recurrent thrombosis. APLAs are considered pathogenic as they play an important role in thrombosis and are not just a serological marker of APS (see Image. Antiphospholipid Syndrome).
A 2-hit thrombosis model is proposed to elucidate thrombus formation in patients with APS. The first hit involves endothelial injury, while the second hit amplifies thrombus formation. Beta-2-glycoprotein-I does not bind unstimulated endothelium in vivo. One hypothesis suggests that when the causes of endothelial injury are unidentified, there may be a disturbance in the redox balance within vascular beds, potentially priming the endothelium. Patients with APS have lower levels of the reduced, protective, and non-immunogenic beta-2 glycoprotein-I. Annexin A2, an endothelial cell surface receptor, is upregulated with oxidative stress. Smoking can lead to endothelial injury and increase pro-thrombotic susceptibilities in patients with lupus anticoagulants.
Plasma nitrite levels are decreased in patients with APS compared to healthy controls. The reduced expression and activity of endothelial nitric oxide synthase result in the generation of peroxynitrite and superoxide. Preclinical models have shown how the domain I of beta-2 glycoprotein-I autoantibodies antagonizes the activity of endothelial nitric oxide synthase with resultant monocyte adhesion and inhibition of nitric oxide–dependent arterial relaxation.
APLAs upregulate tissue factor expression through some intracellular signaling pathways after binding the anti-β2GPI autoantibodies on the surface of monocytes and multiprotein complexes of endothelial cells. Autoantibodies from patients with APS disrupt the mitochondrial function of neutrophils and monocytes and increase the production of reactive oxygen species, resulting in the subsequent expression of tissue factors.[8] Complement activation and inhibition of fibrinolysis by APLAs have been established.
Intraplacental thrombosis, complement pathway activation, interference with trophoblast growth and differentiation, impaired trophoblastic invasion, and effects on hormone production play a role in APS-associated pregnancy loss. In a study involving almost 600 pregnant women with fetal loss after 20 weeks, 9.6% were positive for 1 or more APLA.[6]
Histopathology
Kidney biopsy of patients with APS having renal involvement demonstrates thrombotic microangiopathy, including the following findings—fibrin thrombi in glomeruli or arterioles without inflammatory cells or immune complexes, fibrous intimal hyperplasia, and focal cortical atrophy. Thyroidization may be present, and these lesions should be evaluated separately from lesions consistent with lupus nephritis.[15]
Skin biopsy from sites of nonhealing ulcerations is typically nonspecific and not always performed but may show small vessel and endothelial proliferation without significant vasculitis. A cardiac biopsy may also reveal small vessel thrombosis if performed. Bronchiolar lavage may show hemosiderin-laden macrophages, and lung biopsy may show capillaritis or microthrombi.[15]
History and Physical
The clinical features vary significantly and can be as mild as asymptomatic APLA positivity or as severe as catastrophic APS. Arterial and venous thrombosis and pregnancy-related complications are the hallmarks of the disease. However, several other organ systems may also be involved (noncriteria manifestations).
Vascular Thrombosis
APS can lead to arterial or venous thrombosis involving any organ system. These clots can be isolated or recurrent and involve blood vessels not commonly associated with other causes of thrombosis, such as upper extremity thrombosis, Budd-Chiari syndrome, and sagittal sinus thrombosis. DVTs are the most common venous involvement and may lead to pulmonary embolism, resulting in pulmonary hypertension. Any other site may involve venous thrombosis, including pelvic, renal, mesenteric, hepatic, portal, axillary, ocular, sagittal, and inferior vena cava.[6]
Arterial thrombosis may involve any sized arteries, from the aorta to small capillaries. The most common arterial manifestation of APS is transient ischemic events (TIEs) or ischemic stroke. The occurrence of TIEs or ischemic stroke in young patients without other risk factors for atherosclerosis should raise suspicion for APS. Other sites for arterial thrombosis may include retinal, brachial, coronary, mesenteric, and peripheral arteries. The occurrence of arterial thrombosis carries a poor prognostic value, given the high risk of recurrence in these cases.
Pregnancy Morbidity
Pregnancy loss is common in patients with APS, especially in the second or third trimester. Although genetic and chromosomal defects are the most common causes of early pregnancy loss (<10 weeks of gestation), they may also occur in patients with APS. As noted above, late gestational loss (>20 weeks) is associated with almost a 10% positivity of one or more APLA.[6] Triple positivity, involving testing positive for lupus anticoagulant, anticardiolipin, and anti-β2GPI antibodies; previous pregnancy loss, history of thrombosis; and SLE are risk factors for adverse pregnancy-related outcomes and pregnancy losses in patients with APS. Besides pregnancy losses, other pregnancy-related complications in patients with APS include preeclampsia, fetal distress, premature birth, intrauterine growth retardation, placental insufficiency, placental abruption, and HELLP syndrome (Hemolysis, Elevated Liver enzymes, Low Platelet count).
Cutaneous Involvement
Several cutaneous manifestations have been reported, although all are non-specific for APS. Livedo reticularis is the most common cutaneous manifestation. However, it can also be observed in the healthy population and other disorders such as SLE, other connective tissue diseases, vasculitides, sepsis, multiple cholesterol emboli, and Sneddon syndrome. Skin ulcerations, especially in lower extremities, ranging from small to large ulcers resembling pyoderma gangrenosum, have been reported in patients with APS. Other cutaneous manifestations include nail-fold infarcts, digital gangrene, superficial thrombophlebitis, and necrotizing purpura.[16]
Valvular Involvement
Cardiac valve involvement is prevalent in patients APS, with some studies indicating a prevalence as high as 80%.[17] Mitral and aortic valves are most commonly involved with thickening, nodules, and vegetations evident on echocardiography. These abnormalities may lead to regurgitation or stenosis.
Hematological Involvement
Thrombocytopenia has been observed in more than 15% of APS cases.[18] Severe thrombocytopenia leading to hemorrhage is rare. A positive Coombs test is frequently observed in patients with APS, although hemolytic anemia is rare.
Neurological Involvement
The most common neurological complications of APS include TIEs and ischemic stroke, which may be recurrent, leading to cognitive dysfunction, seizures, and multi-infarct dementia. Blindness secondary to the retinal artery or vein occlusion can also occur. Sudden deafness secondary to sensorineural hearing loss has been reported.
Cardiac Involvement
Myocardial infarction and cardiac emboli are possible complications of APS. A non-ST segment elevation myocardial infarction with a normal coronary angiogram and abnormal cardiac magnetic resonance imaging, such as transmural or subendocardial late gadolinium enhancement, T2 abnormalities, or perfusion abnormalities, suggests APS.[15]
Pulmonary Involvement
Pulmonary artery thromboembolism from DVT is common and may lead to pulmonary hypertension.[19] Diffuse pulmonary hemorrhage resulting from pulmonary capillaritis has been reported.
Renal Involvement
Hypertension, proteinuria, and renal failure secondary to thrombotic microangiopathy are the classic renal manifestations of APS, although this is not specific. Other renal manifestations reported include renal artery thrombosis leading to refractory hypertension and focal cortical atrophy.
Catastrophic Antiphospholipid Syndrome
Catastrophic antiphospholipid syndrome (CAPS) is a rare but life-threatening complication of APS, with fewer than 1% of patients with APS developing CAPS. Mortality rates are high (48%), especially in patients with SLE or those with cardiac, pulmonary, renal, and splenic involvement. CAPS is characterized by thrombosis in multiple organs over a short period, typically spanning a few days. Small- and medium-sized arteries are most frequently involved. Clinical presentation varies depending on the organ involved and may include peripheral thrombosis, such as deep vein thrombosis, femoral artery thrombosis, or radial artery thrombosis; pulmonary complications, such as acute respiratory distress syndrome, pulmonary embolism, and pulmonary hemorrhage; renal manifestations, such as thrombotic microangiopathy and renal failure; cutaneous symptoms, such as livedo reticularis, digital ischemia, gangrene, and skin ulcerations; cerebral manifestations, such as ischemic stroke and encephalopathy; cardiac complications, such as valve lesions, myocardial infarction, and heart failure; hematological issues, such as thrombocytopenia; and gastrointestinal involvement, such as bowel infarction.[20]
The most common precipitating factors for CAPS are stopping anticoagulation in patients diagnosed with APS, infections, and surgical procedures. Prompt management of infections and minimization of no anticoagulation (or using low-dose anticoagulation) perioperatively are recommended.[21]
Preliminary criteria for the classification of CAPS were published in 2003.[22] The 4 criteria are the following:
- Involvement of 3 or more organs, systems, or tissues
- Manifestations developing simultaneously or within less than 1 week
- Histopathological confirmation of small vessel occlusion in at least 1 organ or tissue
- Laboratory confirmation of the presence of APLA
The presence of all 4 criteria can classify definite CAPS, whereas probable CAPS can be classified if 3 criteria are present and the fourth is incompletely fulfilled.
Evaluation
In addition to clinical criteria, diagnosing APS requires lupus anticoagulant or moderate-to-high titers of IgG or IgM anticardiolipin or anti-β2GPI antibodies. The criteria also require a repeat APLA test to be positive 12 weeks after the initial positive test to exclude clinically unimportant or transient antibodies. If that duration is less than 12 weeks, or the gap between two separate clinical manifestations and positive laboratory tests is more than 5 years, the diagnosis of APS is questionable.[23] Please see StatPearls' companion resource, "Antiphospholipid Antibody Testing," for further information.
Lupus Anticoagulant Test
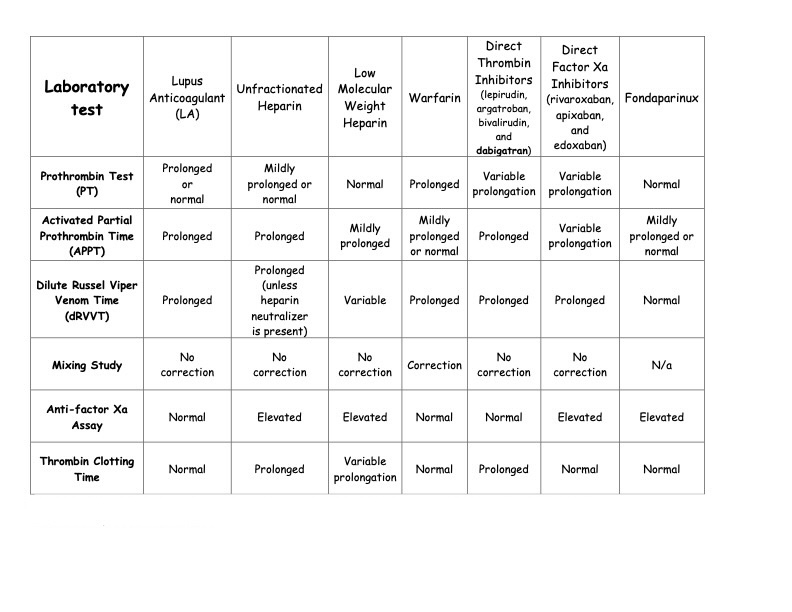
A positive lupus anticoagulant test is the strongest predictor of adverse pregnancy-related events. This test is more specific but less sensitive than anticardiolipin antibodies in predicting thrombosis. A positive lupus anticoagulant test is observed in 20% of patients with anticardiolipin antibodies, and anticardiolipin antibodies are observed in 80% of patients with a positive lupus anticoagulant test (see Table. Effect of Lupus Anticoagulant and Anticoagulants on Laboratory Testing).
A false-positive syphilis test does not meet the criteria for diagnosing APS. However, it is advisable to assess for APLAs in patients with previous thrombotic or adverse pregnancy-related events. The presence of a lupus anticoagulant indicates the presence of an in vitro coagulation inhibitor of phospholipid-dependent coagulation reactions. This inhibitor does not react directly with coagulation factors and is not associated with bleeding complications. False-positive and false-negative results can be observed in patients on heparin or warfarin.
The lupus anticoagulant test involves 4 steps as follows:
- Initial screening, typically involving activated partial thromboplastin time or dilute Russell viper venom time, shows prolonged phospholipid-dependent clot formation.
- The prolonged screening test persists even after mixing the patient's plasma with normal platelet-poor plasma. This lack of correction suggests that the prolonged clotting time is not due to coagulation factor deficiencies, such as hemophilia, which is corrected with normal plasma.
- The prolonged screening test is corrected or improved upon the addition of excess phospholipid, which indicates phospholipid dependency.
- Exclusion of other inhibitors.
Anticardiolipin and Anti-Beta-2-Glycoprotein-I Antibodies
Anticardiolipin antibodies and anti-β2GPI antibodies are assessed by ELISA, and common assays include tests for IgG and IgM isotypes. IgG antibodies correlate better with clinical manifestations compared to IgM or IgA antibodies. Titers of more than 40 IgG units are associated with thrombotic events, whereas lower titers have a less proven association with thrombotic events.
Other Laboratory Findings
Thrombocytopenia or anemia can be frequently observed in patients with APS. Renal failure and proteinuria may indicate renal involvement with thrombotic microangiopathy. The erythrocyte sedimentation rate may be high during the acute thrombotic event. However, markers of inflammation are typically normal otherwise. Patients with SLE may have positive serologies specific for SLE, such as antinuclear, anti-double-stranded DNA, or anti-smith. Hypocomplementemia is not typically observed in patients with APS, and its presence with renal involvement may indicate lupus nephritis.
Notably, positive antinuclear and even anti-double–stranded DNA antibodies are frequently observed in primary APS without associated SLE, and the presence of these antibodies alone does not imply a diagnosis of SLE in patients without any clinical features of lupus. Patients with multiple thrombotic events or pregnancy losses should also be assessed for other hypercoagulable states, such as hyperhomocysteinemia, factor V Leiden and prothrombin mutations, deficiency of protein C, protein S, or antithrombin III, when indicated.
Classification Criteria
The initial classification criteria, known as the Sapporo criteria, were published in 1999 and updated in 2006.[23] The revised Sapporo classification criteria for APS require at least one positive laboratory and one positive clinical criterion.
Clinical Criteria
One of the following clinical findings should be confirmed to diagnose antiphospholipid antibody syndrome.
Vascular Thrombosis
- One or more events of arterial, venous, or small-vessel thrombosis of any organ. Thrombosis must be objectively confirmed with appropriate imaging or histopathology. For histopathology, thrombosis is typically present without significant vessel wall inflammation.
- A past thrombotic episode can be included as a criterion as long as it was appropriately confirmed by appropriate diagnostic means and there was no other cause of thrombosis.
- Superficial venous thrombosis is not included as a criterion.
Pregnancy Morbidity
- One or more unexplained fetal deaths of the morphologically normal fetuses, confirmed by ultrasound or direct examination, at or beyond 10 weeks of gestation.
- One or more premature births of morphologically normal neonates before the 34th week of gestation. Prematurity must be secondary to eclampsia, severe preeclampsia, or placental insufficiency.
- Three or more consecutive spontaneous abortions before the 10th week of gestation after ruling out any anatomical or hormonal abnormalities in the mother and parental chromosomal causes.
Laboratory Criteria
One of the following laboratory findings should be confirmed to diagnose antiphospholipid antibody syndrome.
- Detection of lupus anticoagulant in plasma on 2 or more occasions, at least 12 weeks apart.
- Detection of IgG or IgM anticardiolipin antibodies in serum or plasma at moderate-to-high titers (more than 40 IgG phospholipid (GPL) or IgM phospholipid (GPM) or GPL/GPM >99th percentile) measured by standard ELISA on 2 or more occasions, at least 12 weeks apart.
- Detection of IgG or IgM anti-β2GPI antibodies in serum or plasma at moderate-to-high titers (>99th percentile) measured by standard ELISA on 2 or more occasions, 12 or more weeks apart.
The updated 2023 American College of Rheumatology/European Alliance of Associations for Rheumatology (EULAR) APS classification criteria include an entry criterion of at least one positive antiphospholipid antibody (APLA) test within 3 years of an APS-associated clinical criterion. These criteria are followed by additional weighted criteria, with a score range of 1 to 7 points each, clustered into 6 clinical domains, such as macrovascular venous thromboembolism, macrovascular arterial thrombosis, microvascular, obstetric, cardiac valve, and hematologic, and 2 laboratory domains, such as lupus anticoagulant tests and IgG/IgM anticardiolipin or IgG/IgM β2GP1b. Patients with at least 3 points each from the clinical and laboratory domains are classified as having APS. Compared to the 2006 revised Sapporo classification criteria, the new APS criteria demonstrate a specificity of 99% versus 86% and a sensitivity of 84% versus 99%, indicating more specificity but less sensitivity.[15] The criteria are quite complex, and a full listing can be found at DOI: 10.1002/art.42624.
Treatment / Management
Thrombosis Management
The EULAR has set forth specific guidelines for various clinical scenarios.[21] Primary thromboprophylaxis is debatable in patients with a positive blood test for APLA but no prior history of thrombotic events or pregnancy-related outcomes. Confirmatory testing of APLA is required at least 12 weeks after the initial testing. Patients with SLE who test positive for APLA are especially at a higher risk of developing thrombotic events, and hydroxychloroquine is recommended for these patients due to its thromboprotective properties.[24] Low-dose aspirin may also be considered. Prophylaxis for other patients with APLA showing high-risk APLA profiles, such as triple positivity with other thrombotic risk factors but no history of thrombotic events, may also be considered for low-dose aspirin.
Primary prevention: High-risk patients have a positive lupus anticoagulant, 2 or 3 positive APLA, or persistently high APLA.[21]
- Patients with a high-risk antiphospholipid profile but no history of thrombotic events should be treated with low-dose aspirin (75-100 mg).
- In patients with SLE and no history of thrombosis or pregnancy complications:
- With a high-risk APLA profile, prophylactic treatment with low-dose aspirin is recommended.
- With a low-risk APLA profile, prophylactic treatment with low-dose aspirin may be considered.
- In nonpregnant women with a history of obstetric APS only (with or without SLE), prophylactic treatment with low-dose aspirin after adequate risk/benefit evaluation is recommended.
Secondary prevention
- Venous thrombosis:
- In patients with definite APS and first venous thrombosis, treatment with warfarin with a target international normalized ratio (INR) of 2.0 to 3.0 is recommended, typically initiated with a heparin bridge.[21][25]
- In patients with unprovoked first venous thrombosis, anticoagulation should be continued in the long term.
- Patients with an unprovoked DVT and lupus anticoagulant positive have a 40% increased risk of recurrent thrombosis if anticoagulation is discontinued. Even if anticoagulation is continued, this patient population has a 22% risk of recurrent thrombosis. For this reason, patients diagnosed with APS and thrombosis indicate lifelong anticoagulation, especially for unprovoked events.
- However, a direct oral anticoagulant (DOAC) may be used in selected individuals with venous (as opposed to arterial) thrombosis and single or double antibody positivity.[26]
- Longer anticoagulation can also be considered in patients with a high-risk APLA profile in repeated measurements or other risk factors for recurrence.
- Arterial thrombosis: Warfarin is generally preferred over DOACs for treating arterial thrombosis. The INR goal for patients with arterial thrombosis is debatable. A goal of 2.0 to 3.0 is primarily used, while some suggest a higher goal of more than 3.0.[27]
- Low-molecular-weight heparin (LMWH) can be used in patients who cannot tolerate warfarin or show no response.
- In patients with recurrent thrombosis despite adequate warfarin, adding aspirin to warfarin, changing to LMWH, or high-intensity anticoagulation with an INR goal of more than 3.0 can be considered.
- Investigation of adherence to warfarin treatment, along with frequent INR testing, should also be considered.
- DOACs could be considered in patients unable to achieve a target INR despite good adherence to warfarin or those with contraindications, such as allergy or intolerance to warfarin.
- If a DOAC is used instead of warfarin for patient-specific reasons, dabigatran may be more effective compared to other DOACs, possibly related to its anti-factor IIa mechanism.[26]
- Elderly patients with stroke and low-titer anticardiolipin antibodies can be treated with low-dose aspirin, as low-titer anticardiolipin antibodies may be incidental and not the cause of the stroke.[26]
- An important consideration is that lupus anticoagulant positivity can cause falsely prolonged activated partial prothrombin time (APPT), prothrombin (PT), and INR times. Proposed methods to avoid this include measuring prothrombin levels or factor X activity, which is unavailable in all laboratories (see Table. Effect of Lupus Anticoagulant and Anticoagulants on Laboratory Testing).
Triple positivity: Recently, several trials have compared the efficacy of rivaroxaban and apixaban to warfarin and found that the DOACs were inferior to warfarin. In fact, several studies were terminated early due to excess thrombotic events in the DOAC group. For this reason, several societies recommend against the use of DOACs diagnosed with APS and recommend warfarin treatment in any patient with triple positivity, such as positive lupus anticoagulant, anticardiolipin, and anti-β2GP1b, or arterial thrombosis and APS.[26] Some go even further and recommend that any patients diagnosed with APS and a thrombotic event, either venous or arterial, should be treated with warfarin over a DOAC.
Pregnancy Management
All pregnant females with positive APLA should be under surveillance during their pregnancy to ensure fetal well-being and avoid maternal complications. Treatment for pregnant females is aimed at reducing the risk of adverse fetal outcomes and is dictated by the clinical scenario. Notably, warfarin is teratogenic and is avoided in pregnancy. Although not definitively proven teratogenic, DOACs are also not used in pregnancy due to a lack of safety data. LMWH or unfractionated heparin can be used; however, LMWH is preferred due to its better bioavailability, longer half-life, convenient once-a-day dosing, and lower risk of thrombocytopenia and osteoporosis. The following are EULAR recommendations for pregnant females.[21]
- For pregnant females with positive APLA but no history of arterial or venous thrombosis:
- First pregnancy: No treatment is indicated
- Account of single pregnancy loss at gestation less than 10 weeks: No treatment is indicated
- A high-risk APLA without a history of thrombosis or pregnancy complication: low-dose aspirin should be considered.
- History of 3 or more pregnancy losses at gestation less than 10 weeks or a pregnancy loss at greater than 10 weeks: Low-dose aspirin in combination with prophylactic dose unfractionated heparin or LMWH throughout pregnancy. Aspirin should be started before conception, and aspirin and heparin or LMWH can be discontinued 6 to 12 weeks postpartum, regardless of the route of delivery. LMWH is most commonly used and should be dose-adjusted throughout pregnancy.
- Postpartum anticoagulation is generally continued for at least 6 weeks post-delivery due to increased thrombotic risk.
- Anticoagulation can be resumed 6 to 12 hours after cesarean delivery or 4 to 6 hours after vaginal birth unless there is a significant risk for significant bleeding. Warfarin and heparin are not contraindicated during breastfeeding, and there is a low risk of hemorrhage.[28]
- History of delivery prior to 34th gestational week due to eclampsia, preeclampsia, or other recognized features of placental insufficiency: Treatment with low-dose aspirin or low-dose aspirin plus prophylactic heparin based on the patient's risk profile.
- History of less well-defined criteria, such as 2 spontaneous miscarriages prior to the 10th gestational week or delivery after the 34th gestational week due to eclampsia or preeclampsia: Treatment with low-dose aspirin or prophylactic heparin depending on the patient's risk profile.
- In women with definite obstetric APS with recurrent pregnancy complications despite combination treatment with low-dose aspirin and prophylactic heparin/LMWH, increasing the heparin dose to a therapeutic dose or adding hydroxychloroquine or low-dose prednisolone in the first trimester may be considered. Using intravenous immunoglobulin (IVIG) might be considered in highly selected cases.
- Although the expected benefit of IVIG is small, the panel agreed that this could be considered in highly selected cases when other treatments have failed.
- In women with a history of thrombotic APS, a combination treatment of low-dose aspirin and heparin at therapeutic dosage during pregnancy is recommended.
- In observational studies, treatment with low-dose aspirin and therapeutic dose heparin was associated with an average of 79% live births.
- Switching treatment from warfarin to therapeutic dose heparin/LMWH is recommended as soon as pregnancy is confirmed, ideally before the sixth gestational week, due to the teratogenic effects of warfarin.
Management of Other Manifestations
The role of anticoagulation has not been established in other non-criteria manifestations of APS. Thrombocytopenia with a platelet count of more than 50,000/mL3 does not require any treatment; however, corticosteroids with or without IVIG or rituximab can be used if platelet counts are less than 50,000/mL3. Splenectomy has also been beneficial in some patients with severe refractory thrombocytopenia.
Renal involvement with thrombotic microangiopathy can be confirmed with a renal biopsy to rule out lupus nephritis, especially in patients with concomitant SLE. Anticoagulation and corticosteroids can be used for thrombotic microangiopathy. Effective treatment for patients with cardiac valve nodules or deformities is unknown. However, anticoagulation is recommended if evidence of embolism or intracardiac thrombus is present.
Catastrophic Antiphospholipid Syndrome
Early diagnosis of CAPS is crucial due to its high mortality rates. Combination therapy with glucocorticoids, heparin, and plasma exchange or IVIG is recommended over single agents as first-line treatment of patients with CAPS. Concurrent treatment of precipitating factors, such as infections, gangrene, or malignancy, is also recommended. In cases of refractory CAPS, therapies such as B-cell depletion, including rituximab and cyclophosphamide, or complement inhibition, including eculizumab, may be considered based on data from case reports.[21]
Follow-up Monitoring
Patients who are otherwise tolerating anticoagulation and have no other systemic autoimmune diseases can be scheduled for outpatient visits once or twice a year. Coagulation studies are conducted before initiating anticoagulation and during therapy to guide dosing. In addition, a biochemistry panel, including renal function tests, and a complete blood count are used to monitor patients. Repeated APLA testing is not indicated unless required for future treatment decisions. Moreover, patients who have symptomatic organ-system involvement should have appropriate evaluations based on their symptoms.
Differential Diagnosis
Thrombosis due to antiphospholipid antibody syndrome must be differentiated from other causes, such as hyperhomocysteinemia, factor V Leiden and prothrombin mutations, or deficiencies of protein C, protein S, or antithrombin III.
APS-associated nephropathy must be differentiated from thrombotic thrombocytopenic purpura (TTP), vasculitis, hemolytic uremic syndrome, malignant hypertension, and lupus nephritis. In these cases, a kidney biopsy is often required to establish a diagnosis.
Prognosis
Some European studies have observed 90% to 94% survival over 10 years. However, morbidity is high in patients with APS, with more than 30% of patients developing permanent organ damage and more than 20% developing severe disability at a 10-year follow-up.[29] Poor prognostic features include CAPS, pulmonary hypertension, nephropathy, central nervous system (CNS) involvement, and gangrene of the extremities.
Overall, the prognosis of both primary and secondary APS is similar, but in the latter, the morbidity may be increased due to any underlying rheumatic or autoimmune disorder. Lupus patients with APLAs carry a higher risk of neuropsychiatric disorders.
APLAs are often elevated with underlying malignancy, and elevation is associated with poor prognosis, with or without thromboses.[4]
Complications
Antiphospholipid antibody syndrome can lead to complications of the affected organs including fetal loss, stroke, pulmonary embolism, pulmonary hypertension, valvular abnormality, acute coronary syndrome, mesenteric thrombosis, or hepatic venoocclusive disease.
Perioperative complications are common in patients with APS due to the added prothrombotic risk posed by the surgery. The anticoagulation strategy should be clearly defined before any surgery in patients with APS to prevent thrombosis.
Consultations
The patients are visited by internists, rheumatologists, hematologists, and obstetricians.
Deterrence and Patient Education
Patients should be educated about the possible complications of APS and be informed about symptoms that warrant medical attention, such as those indicative of TIA. Healthcare professionals should clearly communicate the medication regimen and emphasize the importance of medication adherence. Patients on warfarin may require frequent INRs and dietary guidance to avoid interference with warfarin.
Pearls and Other Issues
Identifying and managing other prothrombotic risk factors, such as hyperlipidemia, smoking, hypertension, and oral contraceptives, are essential in patients with APS.
Enhancing Healthcare Team Outcomes
Antiphospholipid antibody syndrome management requires an interprofessional team approach involving multiple specialties. Primary care physicians play the most critical role in identifying patients with APS. Hematologists and rheumatologists play a crucial role in diagnosing, managing, and follow-up. The involvement of other specialties, such as neurology, nephrology, cardiology, and dermatology, may be necessary if the specific organ system is involved. Anticoagulation clinics can play a significant role in monitoring therapeutic warfarin levels and INR with close follow-up.
Given the complexity and variety of underlying conditions leading to antiphospholipid antibody syndrome, all interprofessional team members must be able to provide input for both diagnosis and subsequent treatment, particularly in cases involving pregnancy, where obstetrics and maternal-fetal medicine specialists should also be consulted.
Pharmacists can assist in managing patients, especially by identifying drug interactions because the metabolism of warfarin is affected by several medications; they can also collaborate with clinicians to ensure appropriate warfarin dosing and monitor INR responses. Specialty-trained nurses can also assume this role. Managing patients with APS requires each team member to be able to voice their concerns if therapy is not producing the desired result so that changes in the patient's treatment regimen can be made without delay. Communication among the interprofessional team members and close patient monitoring is vital in managing APS.