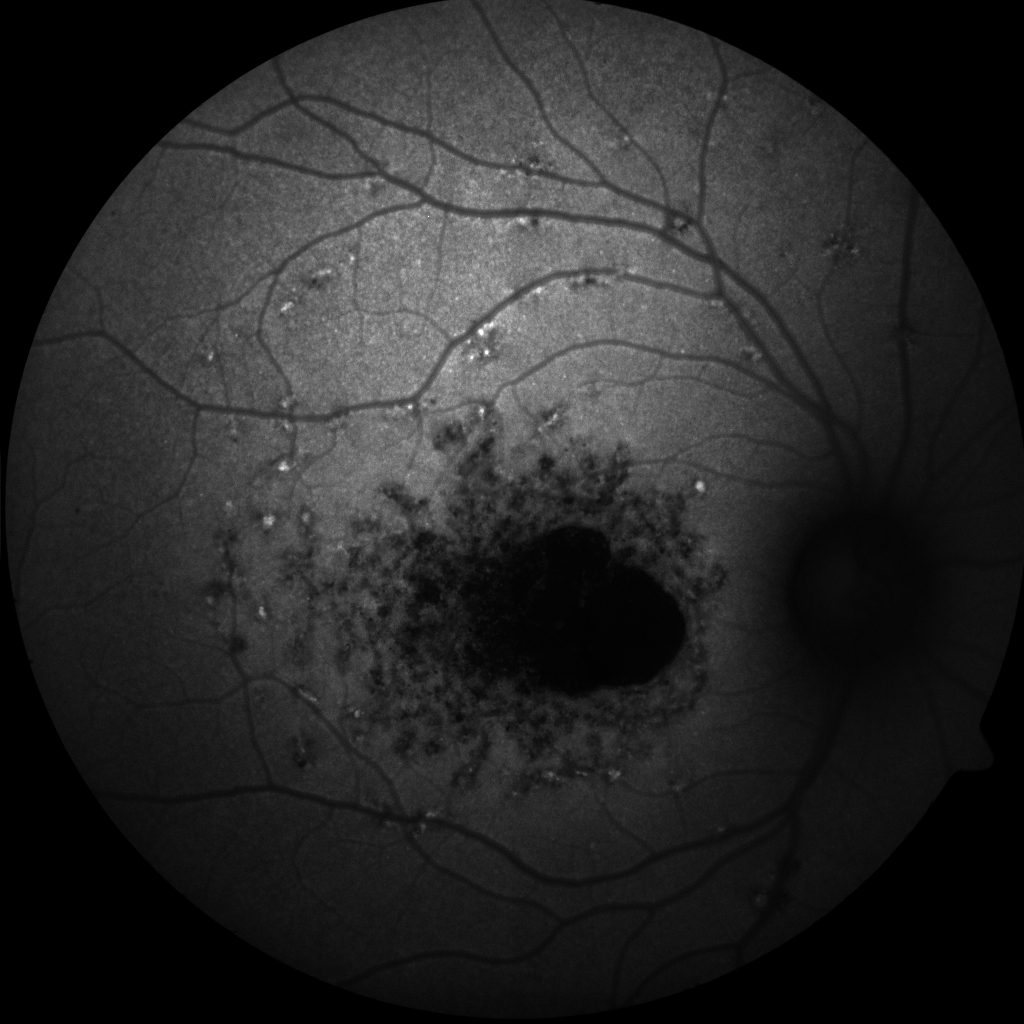
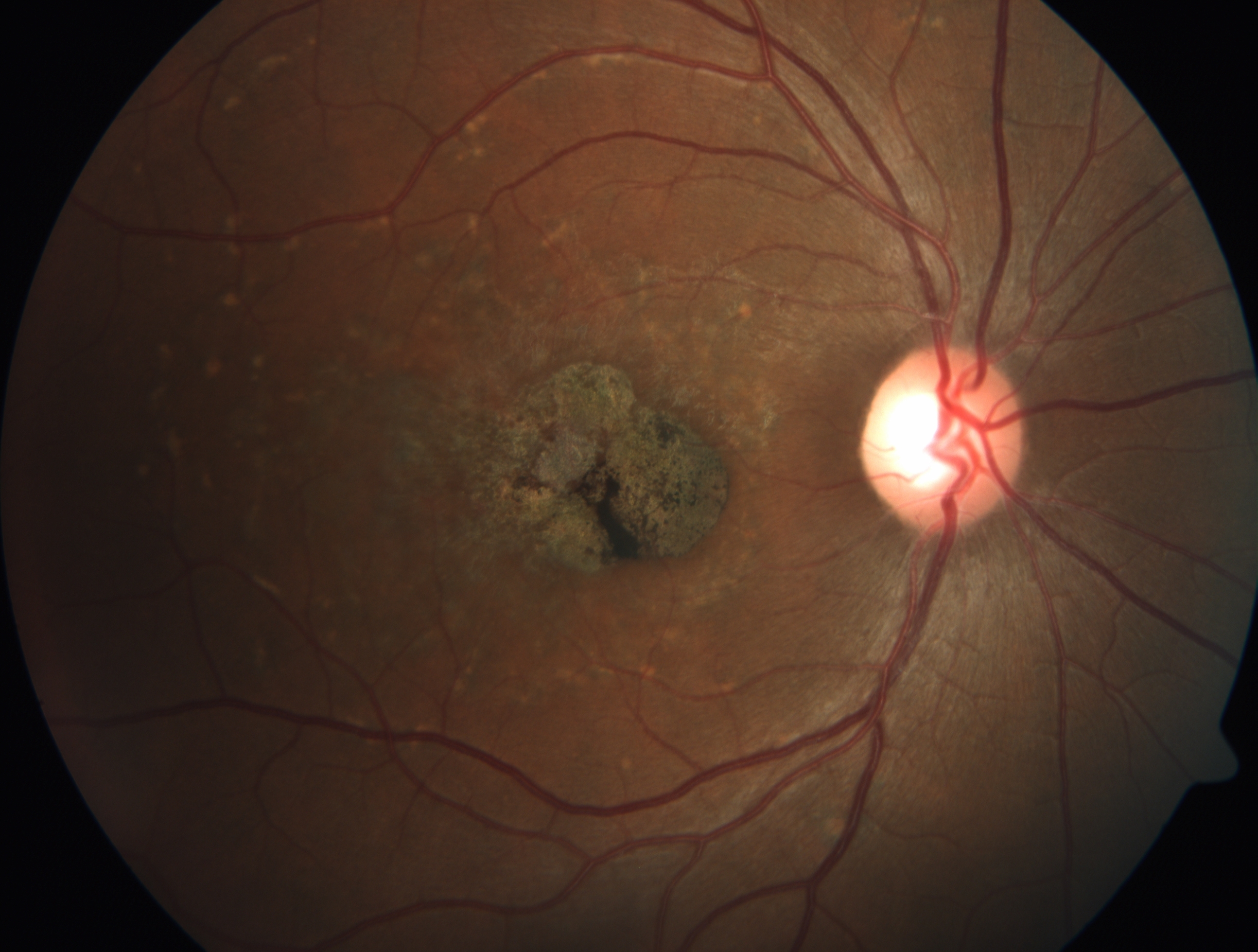
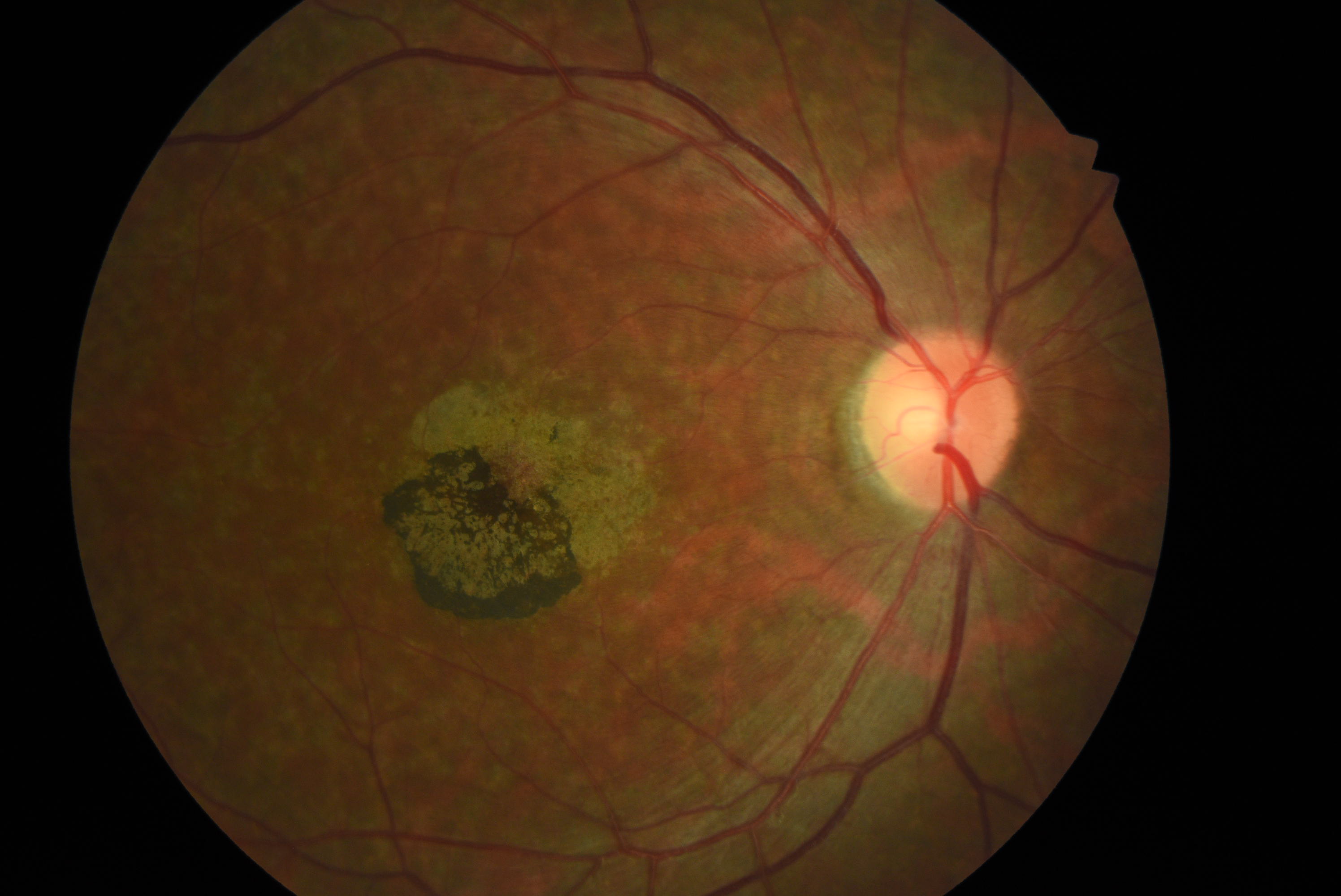
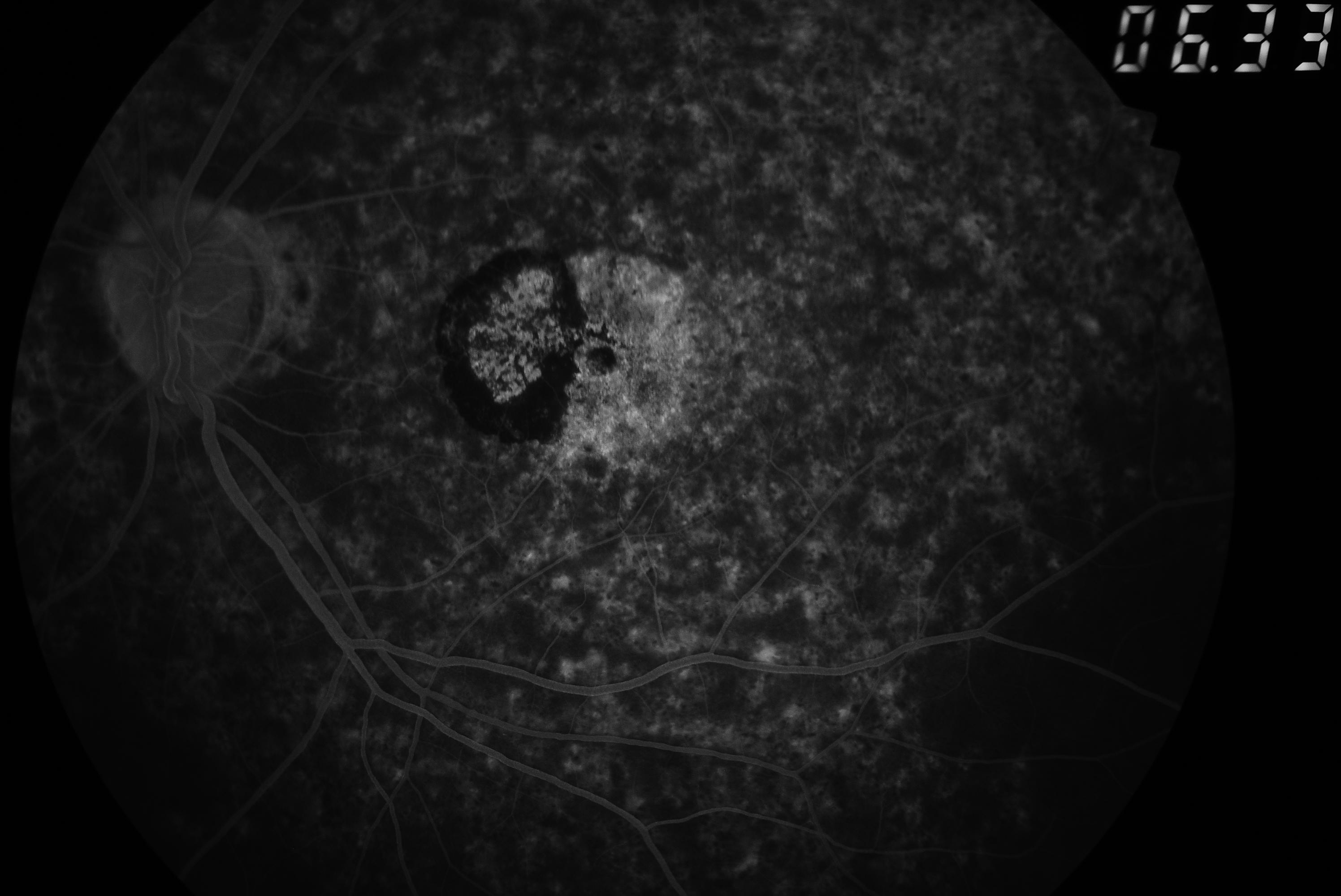
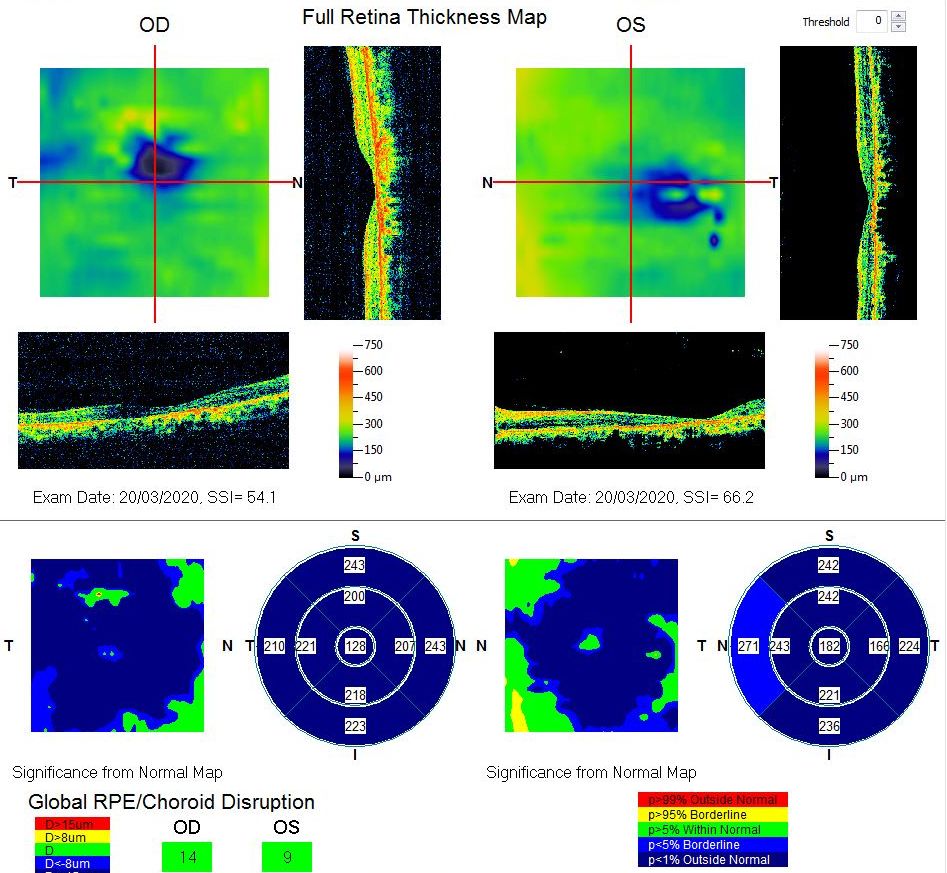
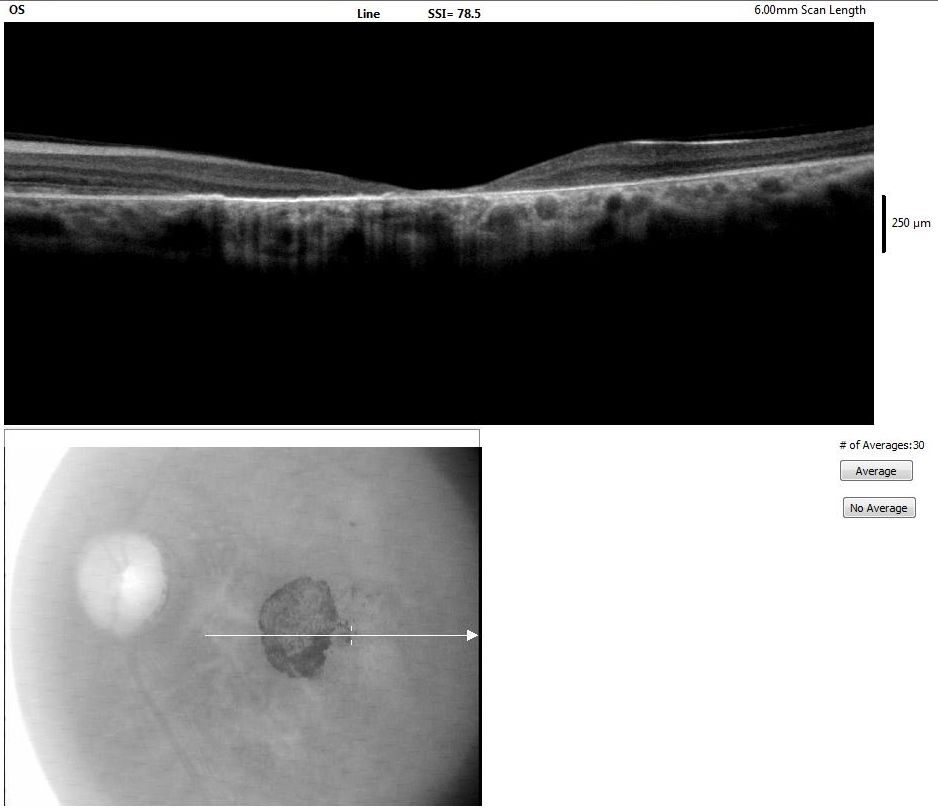
[1]
Strauss RW, Ho A, Muñoz B, Cideciyan AV, Sahel JA, Sunness JS, Birch DG, Bernstein PS, Michaelides M, Traboulsi EI, Zrenner E, Sadda S, Ervin AM, West S, Scholl HP, Progression of Stargardt Disease Study Group. The Natural History of the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Studies: Design and Baseline Characteristics: ProgStar Report No. 1. Ophthalmology. 2016 Apr:123(4):817-28. doi: 10.1016/j.ophtha.2015.12.009. Epub 2016 Jan 16
[PubMed PMID: 26786511]
[2]
Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. The British journal of ophthalmology. 2017 Jan:101(1):25-30. doi: 10.1136/bjophthalmol-2016-308823. Epub 2016 Aug 4
[PubMed PMID: 27491360]
Level 3 (low-level) evidence
[3]
Al-Khuzaei S, Shah M, Foster CR, Yu J, Broadgate S, Halford S, Downes SM. The role of multimodal imaging and vision function testing in ABCA4-related retinopathies and their relevance to future therapeutic interventions. Therapeutic advances in ophthalmology. 2021 Jan-Dec:13():25158414211056384. doi: 10.1177/25158414211056384. Epub 2021 Dec 19
[PubMed PMID: 34988368]
Level 3 (low-level) evidence
[4]
Glazer LC, Dryja TP. Understanding the etiology of Stargardt's disease. Ophthalmology clinics of North America. 2002 Mar:15(1):93-100, viii
[PubMed PMID: 12064087]
Level 3 (low-level) evidence
[5]
Sheffield VC, Stone EM. Genomics and the eye. The New England journal of medicine. 2011 May 19:364(20):1932-42. doi: 10.1056/NEJMra1012354. Epub
[PubMed PMID: 21591945]
[6]
Al-Khuzaei S, Broadgate S, Foster CR, Shah M, Yu J, Downes SM, Halford S. An Overview of the Genetics of ABCA4 Retinopathies, an Evolving Story. Genes. 2021 Aug 13:12(8):. doi: 10.3390/genes12081241. Epub 2021 Aug 13
[PubMed PMID: 34440414]
Level 3 (low-level) evidence
[7]
Tanaka K, Lee W, Zernant J, Schuerch K, Ciccone L, Tsang SH, Sparrow JR, Allikmets R. The Rapid-Onset Chorioretinopathy Phenotype of ABCA4 Disease. Ophthalmology. 2018 Jan:125(1):89-99. doi: 10.1016/j.ophtha.2017.07.019. Epub 2017 Sep 22
[PubMed PMID: 28947085]
[8]
Lee W, Zernant J, Nagasaki T, Tsang SH, Allikmets R. Deep Scleral Exposure: A Degenerative Outcome of End-Stage Stargardt Disease. American journal of ophthalmology. 2018 Nov:195():16-25. doi: 10.1016/j.ajo.2018.07.018. Epub 2018 Jul 26
[PubMed PMID: 30055151]
[9]
Stone EM, Nichols BE, Kimura AE, Weingeist TA, Drack A, Sheffield VC. Clinical features of a Stargardt-like dominant progressive macular dystrophy with genetic linkage to chromosome 6q. Archives of ophthalmology (Chicago, Ill. : 1960). 1994 Jun:112(6):765-72
[PubMed PMID: 8002834]
[10]
Kniazeva M, Chiang MF, Morgan B, Anduze AL, Zack DJ, Han M, Zhang K. A new locus for autosomal dominant stargardt-like disease maps to chromosome 4. American journal of human genetics. 1999 May:64(5):1394-9
[PubMed PMID: 10205271]
[11]
Zhang K, Bither PP, Park R, Donoso LA, Seidman JG, Seidman CE. A dominant Stargardt's macular dystrophy locus maps to chromosome 13q34. Archives of ophthalmology (Chicago, Ill. : 1960). 1994 Jun:112(6):759-64
[PubMed PMID: 8002833]
[12]
Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, Li Y, Metzker ML, Allikmets R, Zack DJ, Kakuk LE, Lagali PS, Wong PW, MacDonald IM, Sieving PA, Figueroa DJ, Austin CP, Gould RJ, Ayyagari R, Petrukhin K. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nature genetics. 2001 Jan:27(1):89-93
[PubMed PMID: 11138005]
[13]
Griesinger IB, Sieving PA, Ayyagari R. Autosomal dominant macular atrophy at 6q14 excludes CORD7 and MCDR1/PBCRA loci. Investigative ophthalmology & visual science. 2000 Jan:41(1):248-55
[PubMed PMID: 10634627]
[15]
Imani S, Cheng J, Shasaltaneh MD, Wei C, Yang L, Fu S, Zou H, Khan MA, Zhang X, Chen H, Zhang D, Duan C, Lv H, Li Y, Chen R, Fu J. Genetic identification and molecular modeling characterization reveal a novel PROM1 mutation in Stargardt4-like macular dystrophy. Oncotarget. 2018 Jan 2:9(1):122-141. doi: 10.18632/oncotarget.22343. Epub 2017 Nov 9
[PubMed PMID: 29416601]
[16]
Lee W, Paavo M, Zernant J, Stong N, Laurente Z, Bearelly S, Nagasaki T, Tsang SH, Goldstein DB, Allikmets R. Modification of the PROM1 disease phenotype by a mutation in ABCA4. Ophthalmic genetics. 2019 Aug:40(4):369-375. doi: 10.1080/13816810.2019.1660382. Epub 2019 Sep 6
[PubMed PMID: 31576780]
[17]
Sajovic J, Meglič A, Volk M, Maver A, Jarc-Vidmar M, Hawlina M, Fakin A. Stargardt-like Clinical Characteristics and Disease Course Associated with Variants in the WDR19 Gene. Genes. 2023 Jan 22:14(2):. doi: 10.3390/genes14020291. Epub 2023 Jan 22
[PubMed PMID: 36833218]
[18]
Spiteri Cornish K, Ho J, Downes S, Scott NW, Bainbridge J, Lois N. The Epidemiology of Stargardt Disease in the United Kingdom. Ophthalmology. Retina. 2017 Nov-Dec:1(6):508-513. doi: 10.1016/j.oret.2017.03.001. Epub 2017 Apr 21
[PubMed PMID: 31047443]
[19]
Runhart EH, Dhooge P, Meester-Smoor M, Pas J, Pott JWR, van Leeuwen R, Kroes HY, Bergen AA, de Jong-Hesse Y, Thiadens AA, van Schooneveld MJ, van Genderen M, Boon C, Klaver C, van den Born LI, Cremers FPM, Hoyng CB. Stargardt disease: monitoring incidence and diagnostic trends in the Netherlands using a nationwide disease registry. Acta ophthalmologica. 2022 Jun:100(4):395-402. doi: 10.1111/aos.14996. Epub 2021 Aug 25
[PubMed PMID: 34431609]
[20]
Molday RS, Zhong M, Quazi F. The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochimica et biophysica acta. 2009 Jul:1791(7):573-83. doi: 10.1016/j.bbalip.2009.02.004. Epub 2009 Feb 20
[PubMed PMID: 19230850]
[21]
Delori FC, Dorey CK, Staurenghi G, Arend O, Goger DG, Weiter JJ. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Investigative ophthalmology & visual science. 1995 Mar:36(3):718-29
[PubMed PMID: 7890502]
[22]
Delori FC, Staurenghi G, Arend O, Dorey CK, Goger DG, Weiter JJ. In vivo measurement of lipofuscin in Stargardt's disease--Fundus flavimaculatus. Investigative ophthalmology & visual science. 1995 Oct:36(11):2327-31
[PubMed PMID: 7558729]
[23]
Eldred GE, Lasky MR. Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature. 1993 Feb 25:361(6414):724-6
[PubMed PMID: 8441466]
[24]
Westerfeld C, Mukai S. Stargardt's disease and the ABCR gene. Seminars in ophthalmology. 2008 Jan-Feb:23(1):59-65. doi: 10.1080/08820530701745249. Epub
[PubMed PMID: 18214793]
[25]
Sparrow JR, Marsiglia M, Allikmets R, Tsang S, Lee W, Duncker T, Zernant J. Flecks in Recessive Stargardt Disease: Short-Wavelength Autofluorescence, Near-Infrared Autofluorescence, and Optical Coherence Tomography. Investigative ophthalmology & visual science. 2015 Jul:56(8):5029-39. doi: 10.1167/iovs.15-16763. Epub
[PubMed PMID: 26230768]
[26]
Bonilha VL, Rayborn ME, Bell BA, Marino MJ, Fishman GA, Hollyfield JG. Retinal Histopathology in Eyes from a Patient with Stargardt disease caused by Compound Heterozygous ABCA4 Mutations. Ophthalmic genetics. 2016 Jun:37(2):150-60. doi: 10.3109/13816810.2014.958861. Epub 2014 Sep 29
[PubMed PMID: 25265374]
[27]
Birnbach CD, Järveläinen M, Possin DE, Milam AH. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology. 1994 Jul:101(7):1211-9
[PubMed PMID: 8035984]
[28]
Cremers FPM, Lee W, Collin RWJ, Allikmets R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Progress in retinal and eye research. 2020 Nov:79():100861. doi: 10.1016/j.preteyeres.2020.100861. Epub 2020 Apr 9
[PubMed PMID: 32278709]
[29]
Tripathy K, Sharma YR, Chawla R, Basu K, Vohra R, Venkatesh P. Triads in Ophthalmology: A Comprehensive Review. Seminars in ophthalmology. 2017:32(2):237-250. doi: 10.3109/08820538.2015.1045150. Epub 2015 Jul 6
[PubMed PMID: 26148300]
[30]
Fujinami K, Zernant J, Chana RK, Wright GA, Tsunoda K, Ozawa Y, Tsubota K, Robson AG, Holder GE, Allikmets R, Michaelides M, Moore AT. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. 2015 Feb:122(2):326-34. doi: 10.1016/j.ophtha.2014.08.012. Epub 2014 Oct 12
[PubMed PMID: 25312043]
[32]
Heath Jeffery RC, Chen FK. Stargardt disease: Multimodal imaging: A review. Clinical & experimental ophthalmology. 2021 Jul:49(5):498-515. doi: 10.1111/ceo.13947. Epub 2021 Jun 1
[PubMed PMID: 34013643]
[33]
van Huet RA, Bax NM, Westeneng-Van Haaften SC, Muhamad M, Zonneveld-Vrieling MN, Hoefsloot LH, Cremers FP, Boon CJ, Klevering BJ, Hoyng CB. Foveal sparing in Stargardt disease. Investigative ophthalmology & visual science. 2014 Oct 16:55(11):7467-78. doi: 10.1167/iovs.13-13825. Epub 2014 Oct 16
[PubMed PMID: 25324290]
[34]
Lambertus S, van Huet RA, Bax NM, Hoefsloot LH, Cremers FP, Boon CJ, Klevering BJ, Hoyng CB. Early-onset stargardt disease: phenotypic and genotypic characteristics. Ophthalmology. 2015 Feb:122(2):335-44. doi: 10.1016/j.ophtha.2014.08.032. Epub 2014 Oct 17
[PubMed PMID: 25444351]
[35]
Fishman GA. Fundus flavimaculatus. A clinical classification. Archives of ophthalmology (Chicago, Ill. : 1960). 1976 Dec:94(12):2061-7
[PubMed PMID: 999551]
[36]
Noble KG, Carr RE. Stargardt's disease and fundus flavimaculatus. Archives of ophthalmology (Chicago, Ill. : 1960). 1979 Jul:97(7):1281-5
[PubMed PMID: 454263]
[37]
Jayasundera T, Rhoades W, Branham K, Niziol LM, Musch DC, Heckenlively JR. Peripapillary dark choroid ring as a helpful diagnostic sign in advanced stargardt disease. American journal of ophthalmology. 2010 Apr:149(4):656-660.e2. doi: 10.1016/j.ajo.2009.11.005. Epub 2010 Feb 6
[PubMed PMID: 20138608]
[38]
Querques G, Leveziel N, Benhamou N, Voigt M, Soubrane G, Souied EH. Analysis of retinal flecks in fundus flavimaculatus using optical coherence tomography. The British journal of ophthalmology. 2006 Sep:90(9):1157-62
[PubMed PMID: 16754647]
[39]
Nõupuu K, Lee W, Zernant J, Tsang SH, Allikmets R. Structural and genetic assessment of the ABCA4-associated optical gap phenotype. Investigative ophthalmology & visual science. 2014 Oct 9:55(11):7217-26. doi: 10.1167/iovs.14-14674. Epub 2014 Oct 9
[PubMed PMID: 25301883]
[40]
Burke TR, Yzer S, Zernant J, Smith RT, Tsang SH, Allikmets R. Abnormality in the external limiting membrane in early Stargardt disease. Ophthalmic genetics. 2013 Mar-Jun:34(1-2):75-7. doi: 10.3109/13816810.2012.707271. Epub 2012 Aug 7
[PubMed PMID: 22871184]
[41]
Pang CE, Suqin Y, Sherman J, Freund KB. New insights into Stargardt disease with multimodal imaging. Ophthalmic surgery, lasers & imaging retina. 2015 Feb:46(2):257-61. doi: 10.3928/23258160-20150213-09. Epub
[PubMed PMID: 25707054]
Level 2 (mid-level) evidence
[42]
Khan KN, Kasilian M, Mahroo OAR, Tanna P, Kalitzeos A, Robson AG, Tsunoda K, Iwata T, Moore AT, Fujinami K, Michaelides M. Early Patterns of Macular Degeneration in ABCA4-Associated Retinopathy. Ophthalmology. 2018 May:125(5):735-746. doi: 10.1016/j.ophtha.2017.11.020. Epub 2018 Jan 6
[PubMed PMID: 29310964]
[43]
Georgiou M, Kane T, Tanna P, Bouzia Z, Singh N, Kalitzeos A, Strauss RW, Fujinami K, Michaelides M. Prospective Cohort Study of Childhood-Onset Stargardt Disease: Fundus Autofluorescence Imaging, Progression, Comparison with Adult-Onset Disease, and Disease Symmetry. American journal of ophthalmology. 2020 Mar:211():159-175. doi: 10.1016/j.ajo.2019.11.008. Epub 2019 Dec 6
[PubMed PMID: 31812472]
[44]
Arrigo A, Grazioli A, Romano F, Aragona E, Bordato A, di Nunzio C, Sperti A, Bandello F, Battaglia Parodi M. Choroidal Patterns in Stargardt Disease: Correlations with Visual Acuity and Disease Progression. Journal of clinical medicine. 2019 Sep 5:8(9):. doi: 10.3390/jcm8091388. Epub 2019 Sep 5
[PubMed PMID: 31491905]
[45]
Cai CX, Light JG, Handa JT. Quantifying the Rate of Ellipsoid Zone Loss in Stargardt Disease. American journal of ophthalmology. 2018 Feb:186():1-9. doi: 10.1016/j.ajo.2017.10.032. Epub 2017 Nov 7
[PubMed PMID: 29126757]
[46]
Piri N, Nesmith BL, Schaal S. Choroidal hyperreflective foci in Stargardt disease shown by spectral-domain optical coherence tomography imaging: correlation with disease severity. JAMA ophthalmology. 2015 Apr:133(4):398-405. doi: 10.1001/jamaophthalmol.2014.5604. Epub
[PubMed PMID: 25590640]
[47]
Fujinami K, Lois N, Mukherjee R, McBain VA, Tsunoda K, Tsubota K, Stone EM, Fitzke FW, Bunce C, Moore AT, Webster AR, Michaelides M. A longitudinal study of Stargardt disease: quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Investigative ophthalmology & visual science. 2013 Dec 17:54(13):8181-90. doi: 10.1167/iovs.13-12104. Epub 2013 Dec 17
[PubMed PMID: 24265018]
[48]
Kuehlewein L, Hariri AH, Ho A, Dustin L, Wolfson Y, Strauss RW, Scholl HP, Sadda SR. COMPARISON OF MANUAL AND SEMIAUTOMATED FUNDUS AUTOFLUORESCENCE ANALYSIS OF MACULAR ATROPHY IN STARGARDT DISEASE PHENOTYPE. Retina (Philadelphia, Pa.). 2016 Jun:36(6):1216-21. doi: 10.1097/IAE.0000000000000870. Epub
[PubMed PMID: 26583307]
[49]
Strauss RW, Muñoz B, Ho A, Jha A, Michaelides M, Mohand-Said S, Cideciyan AV, Birch D, Hariri AH, Nittala MG, Sadda S, Scholl HPN, ProgStar Study Group. Incidence of Atrophic Lesions in Stargardt Disease in the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Study: Report No. 5. JAMA ophthalmology. 2017 Jul 1:135(7):687-695. doi: 10.1001/jamaophthalmol.2017.1121. Epub
[PubMed PMID: 28542697]
[50]
Klufas MA, Tsui I, Sadda SR, Hosseini H, Schwartz SD. ULTRAWIDEFIELD AUTOFLUORESENCE IN ABCA4 STARGARDT DISEASE. Retina (Philadelphia, Pa.). 2018 Feb:38(2):403-415. doi: 10.1097/IAE.0000000000001567. Epub
[PubMed PMID: 28248825]
[51]
Delori F, Greenberg JP, Woods RL, Fischer J, Duncker T, Sparrow J, Smith RT. Quantitative measurements of autofluorescence with the scanning laser ophthalmoscope. Investigative ophthalmology & visual science. 2011 Dec 9:52(13):9379-90. doi: 10.1167/iovs.11-8319. Epub 2011 Dec 9
[PubMed PMID: 22016060]
[52]
Song H, Rossi EA, Latchney L, Bessette A, Stone E, Hunter JJ, Williams DR, Chung M. Cone and rod loss in Stargardt disease revealed by adaptive optics scanning light ophthalmoscopy. JAMA ophthalmology. 2015 Oct:133(10):1198-203. doi: 10.1001/jamaophthalmol.2015.2443. Epub
[PubMed PMID: 26247787]
[54]
Schroeder M, Kjellström U. Full-field ERG as a predictor of the natural course of ABCA4-associated retinal degenerations. Molecular vision. 2018:24():1-16
[PubMed PMID: 29386879]
[55]
Schönbach EM, Wolfson Y, Strauss RW, Ibrahim MA, Kong X, Muñoz B, Birch DG, Cideciyan AV, Hahn GA, Nittala M, Sunness JS, Sadda SR, West SK, Scholl HPN, ProgStar Study Group. Macular Sensitivity Measured With Microperimetry in Stargardt Disease in the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Study: Report No. 7. JAMA ophthalmology. 2017 Jul 1:135(7):696-703. doi: 10.1001/jamaophthalmol.2017.1162. Epub
[PubMed PMID: 28542693]
[56]
Kong X, Ibrahim-Ahmed M, Bittencourt MG, Strauss RW, Birch DG, Cideciyan AV, Ervin AM, Ho A, Sunness JS, Audo IS, Michaelides M, Zrenner E, Sadda S, Ip MS, West S, Scholl HPN, SMART Study Group. Longitudinal Changes in Scotopic and Mesopic Macular Function as Assessed with Microperimetry in Patients With Stargardt Disease: SMART Study Report No. 2. American journal of ophthalmology. 2022 Apr:236():32-44. doi: 10.1016/j.ajo.2021.10.014. Epub 2021 Oct 23
[PubMed PMID: 34695402]
[57]
Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Archives of ophthalmology (Chicago, Ill. : 1960). 2001 Mar:119(3):359-69
[PubMed PMID: 11231769]
[58]
Fujinami K, Lois N, Davidson AE, Mackay DS, Hogg CR, Stone EM, Tsunoda K, Tsubota K, Bunce C, Robson AG, Moore AT, Webster AR, Holder GE, Michaelides M. A longitudinal study of stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. American journal of ophthalmology. 2013 Jun:155(6):1075-1088.e13. doi: 10.1016/j.ajo.2013.01.018. Epub 2013 Mar 15
[PubMed PMID: 23499370]
[59]
Kretschmann U, Seeliger MW, Ruether K, Usui T, Apfelstedt-Sylla E, Zrenner E. Multifocal electroretinography in patients with Stargardt's macular dystrophy. The British journal of ophthalmology. 1998 Mar:82(3):267-75
[PubMed PMID: 9602623]
[60]
Stavrou P, Good PA, Misson GP, Kritzinger EE. Electrophysiological findings in Stargardt's-fundus flavimaculatus disease. Eye (London, England). 1998:12 ( Pt 6)():953-8
[PubMed PMID: 10325994]
[61]
Huang D, Heath Jeffery RC, Aung-Htut MT, McLenachan S, Fletcher S, Wilton SD, Chen FK. Stargardt disease and progress in therapeutic strategies. Ophthalmic genetics. 2022 Feb:43(1):1-26. doi: 10.1080/13816810.2021.1966053. Epub 2021 Aug 29
[PubMed PMID: 34455905]
[62]
Kubota R, Boman NL, David R, Mallikaarjun S, Patil S, Birch D. Safety and effect on rod function of ACU-4429, a novel small-molecule visual cycle modulator. Retina (Philadelphia, Pa.). 2012 Jan:32(1):183-8. doi: 10.1097/IAE.0b013e318217369e. Epub
[PubMed PMID: 21519291]
[63]
Kubota R, Al-Fayoumi S, Mallikaarjun S, Patil S, Bavik C, Chandler JW. Phase 1, dose-ranging study of emixustat hydrochloride (ACU-4429), a novel visual cycle modulator, in healthy volunteers. Retina (Philadelphia, Pa.). 2014 Mar:34(3):603-9. doi: 10.1097/01.iae.0000434565.80060.f8. Epub
[PubMed PMID: 24056528]
[64]
Kubota R, Birch DG, Gregory JK, Koester JM. Randomised study evaluating the pharmacodynamics of emixustat hydrochloride in subjects with macular atrophy secondary to Stargardt disease. The British journal of ophthalmology. 2022 Mar:106(3):403-408. doi: 10.1136/bjophthalmol-2020-317712. Epub 2020 Nov 19
[PubMed PMID: 33214244]
Level 1 (high-level) evidence
[65]
Charbel Issa P, Barnard AR, Herrmann P, Washington I, MacLaren RE. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proceedings of the National Academy of Sciences of the United States of America. 2015 Jul 7:112(27):8415-20. doi: 10.1073/pnas.1506960112. Epub 2015 Jun 23
[PubMed PMID: 26106163]
[66]
Mata NL, Lichter JB, Vogel R, Han Y, Bui TV, Singerman LJ. Investigation of oral fenretinide for treatment of geographic atrophy in age-related macular degeneration. Retina (Philadelphia, Pa.). 2013 Mar:33(3):498-507. doi: 10.1097/IAE.0b013e318265801d. Epub
[PubMed PMID: 23023528]
[67]
Federspiel CA, Bertelsen M, Kessel L. Vitamin A in Stargardt disease-an evidence-based update. Ophthalmic genetics. 2018 Oct:39(5):555-559. doi: 10.1080/13816810.2018.1488174. Epub 2018 Jun 25
[PubMed PMID: 29939824]
[68]
Trapani I, Toriello E, de Simone S, Colella P, Iodice C, Polishchuk EV, Sommella A, Colecchi L, Rossi S, Simonelli F, Giunti M, Bacci ML, Polishchuk RS, Auricchio A. Improved dual AAV vectors with reduced expression of truncated proteins are safe and effective in the retina of a mouse model of Stargardt disease. Human molecular genetics. 2015 Dec 1:24(23):6811-25. doi: 10.1093/hmg/ddv386. Epub 2015 Sep 29
[PubMed PMID: 26420842]
[69]
Binley K, Widdowson P, Loader J, Kelleher M, Iqball S, Ferrige G, de Belin J, Carlucci M, Angell-Manning D, Hurst F, Ellis S, Miskin J, Fernandes A, Wong P, Allikmets R, Bergstrom C, Aaberg T, Yan J, Kong J, Gouras P, Prefontaine A, Vezina M, Bussieres M, Naylor S, Mitrophanous KA. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: safety and biodistribution of StarGen for Stargardt disease. Investigative ophthalmology & visual science. 2013 Jun 12:54(6):4061-71. doi: 10.1167/iovs.13-11871. Epub 2013 Jun 12
[PubMed PMID: 23620430]
[70]
Ricca AM, Han IC, Sohn EH. Stargardt disease masquerades. Current opinion in ophthalmology. 2021 May 1:32(3):214-224. doi: 10.1097/ICU.0000000000000750. Epub
[PubMed PMID: 33653979]
Level 3 (low-level) evidence
[71]
Marmor MF, Byers B. Pattern dystrophy of the pigment epithelium. American journal of ophthalmology. 1977 Jul:84(1):32-44
[PubMed PMID: 900215]
[72]
Francis PJ, Schultz DW, Gregory AM, Schain MB, Barra R, Majewski J, Ott J, Acott T, Weleber RG, Klein ML. Genetic and phenotypic heterogeneity in pattern dystrophy. The British journal of ophthalmology. 2005 Sep:89(9):1115-9
[PubMed PMID: 16113362]
[73]
Kuper WFE, Talsma HE, van Schooneveld MJ, Pott JWR, Huijgen BCH, de Wit GC, van Hasselt PM, van Genderen MM. Recognizing differentiating clinical signs of CLN3 disease (Batten disease) at presentation. Acta ophthalmologica. 2021 Jun:99(4):397-404. doi: 10.1111/aos.14630. Epub 2020 Oct 18
[PubMed PMID: 33073538]
[74]
Tripathy K, Sarma B, Mazumdar S. Outer retinal tubulation and inner retinal pseudocysts in a patient with maternally inherited diabetes and deafness evaluated with optical coherence tomography angiogram. Indian journal of ophthalmology. 2020 Jan:68(1):250-253. doi: 10.4103/ijo.IJO_577_19. Epub
[PubMed PMID: 31856543]
[75]
Raja MS, Goldsmith C, Burton BJ. Outer retinal tubulations in maternally inherited diabetes and deafness (MIDD)-associated macular dystrophy. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2013 Sep:251(9):2265-7. doi: 10.1007/s00417-012-2217-z. Epub 2013 Jan 12
[PubMed PMID: 23314478]
[76]
Westeneng-van Haaften SC, Boon CJ, Cremers FP, Hoefsloot LH, den Hollander AI, Hoyng CB. Clinical and genetic characteristics of late-onset Stargardt's disease. Ophthalmology. 2012 Jun:119(6):1199-210. doi: 10.1016/j.ophtha.2012.01.005. Epub 2012 Mar 24
[PubMed PMID: 22449572]
[77]
Grandinetti AA, Portella E, Arana J, Iskorostenski NT. Subretinal fibrosis in Stargardt's disease: case report. Arquivos brasileiros de oftalmologia. 2011 Nov-Dec:74(6):449-51
[PubMed PMID: 22331122]
Level 3 (low-level) evidence
[78]
Gass JD, Hummer J. Focal retinal pigment epithelial dysplasia associated with fundus flavimaculatus. Retina (Philadelphia, Pa.). 1999:19(4):297-301
[PubMed PMID: 10458294]
[79]
Rossi S, Testa F, Attanasio M, Orrico A, de Benedictis A, Corte MD, Simonelli F. Subretinal Fibrosis in Stargardt's Disease with Fundus Flavimaculatus and ABCA4 Gene Mutation. Case reports in ophthalmology. 2012 Sep:3(3):410-7. doi: 10.1159/000345415. Epub 2012 Dec 20
[PubMed PMID: 23341817]
Level 3 (low-level) evidence
[80]
Paterick TE, Patel N, Tajik AJ, Chandrasekaran K. Improving health outcomes through patient education and partnerships with patients. Proceedings (Baylor University. Medical Center). 2017 Jan:30(1):112-113
[PubMed PMID: 28152110]