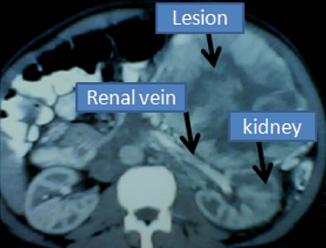
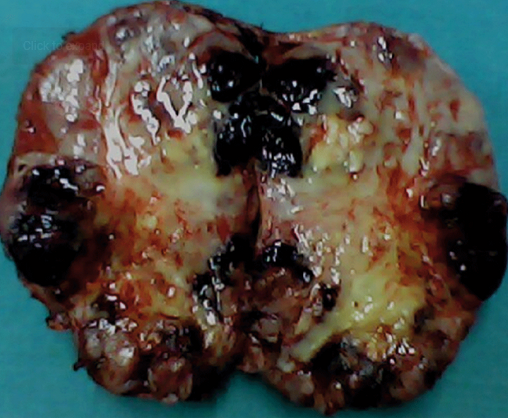
[1]
Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K, Young WF Jr, Endocrine Society. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. The Journal of clinical endocrinology and metabolism. 2014 Jun:99(6):1915-42. doi: 10.1210/jc.2014-1498. Epub
[PubMed PMID: 24893135]
Level 1 (high-level) evidence
[2]
Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension (Dallas, Tex. : 1979). 1997 May:29(5):1133-9
[PubMed PMID: 9149678]
[3]
Neumann HP, Young WF Jr, Krauss T, Bayley JP, Schiavi F, Opocher G, Boedeker CC, Tirosh A, Castinetti F, Ruf J, Beltsevich D, Walz M, Groeben HT, von Dobschuetz E, Gimm O, Wohllk N, Pfeifer M, Lourenço DM Jr, Peczkowska M, Patocs A, Ngeow J, Makay Ö, Shah NS, Tischler A, Leijon H, Pennelli G, Villar Gómez de Las Heras K, Links TP, Bausch B, Eng C. 65 YEARS OF THE DOUBLE HELIX: Genetics informs precision practice in the diagnosis and management of pheochromocytoma. Endocrine-related cancer. 2018 Aug:25(8):T201-T219. doi: 10.1530/ERC-18-0085. Epub 2018 May 24
[PubMed PMID: 29794110]
[4]
Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. American journal of surgery. 2000 Mar:179(3):212-5
[PubMed PMID: 10827323]
[5]
Ariton M, Juan CS, AvRuskin TW. Pheochromocytoma: clinical observations from a Brooklyn tertiary hospital. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2000 May-Jun:6(3):249-52
[PubMed PMID: 11421540]
[6]
Mariani-Costantini R, Shen Y, Cheng L. Biochemical Diagnosis of Pheochromocytoma and Paraganglioma. Paraganglioma: A Multidisciplinary Approach. 2019 Jul 2:():
[PubMed PMID: 31294941]
[7]
Pacak K. Preoperative management of the pheochromocytoma patient. The Journal of clinical endocrinology and metabolism. 2007 Nov:92(11):4069-79
[PubMed PMID: 17989126]
[8]
Gupta G, Pacak K, AACE Adrenal Scientific Committee. PRECISION MEDICINE: AN UPDATE ON GENOTYPE/BIOCHEMICAL PHENOTYPE RELATIONSHIPS IN PHEOCHROMOCYTOMA/PARAGANGLIOMA PATIENTS. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2017 Jun:23(6):690-704. doi: 10.4158/EP161718.RA. Epub 2017 Mar 23
[PubMed PMID: 28332883]
[9]
Sundaresan PR, Banerjee SP. Differential regulation of beta-adrenergic receptor-coupled adenylate cyclase by thyroid hormones in rat liver and heart: possible role of corticosteroids. Hormone research. 1987:27(2):109-18
[PubMed PMID: 2820855]
[10]
Motulsky HJ, Insel PA. Adrenergic receptors in man: direct identification, physiologic regulation, and clinical alterations. The New England journal of medicine. 1982 Jul 1:307(1):18-29
[PubMed PMID: 6123082]
[11]
Ciaraldi TP, Marinetti GV. Hormone action at the membrane level. VIII. Adrenergic receptors in rat heart and adipocytes and their modulation by thyroxine. Biochimica et biophysica acta. 1978 Jul 3:541(3):334-46
[PubMed PMID: 149563]
[12]
Nazari MA, Rosenblum JS, Haigney MC, Rosing DR, Pacak K. Pathophysiology and Acute Management of Tachyarrhythmias in Pheochromocytoma: JACC Review Topic of the Week. Journal of the American College of Cardiology. 2020 Jul 28:76(4):451-464. doi: 10.1016/j.jacc.2020.04.080. Epub
[PubMed PMID: 32703516]
[13]
Kushnir A, Marks AR. The ryanodine receptor in cardiac physiology and disease. Advances in pharmacology (San Diego, Calif.). 2010:59():1-30. doi: 10.1016/S1054-3589(10)59001-X. Epub
[PubMed PMID: 20933197]
Level 3 (low-level) evidence
[14]
Wakabayashi T. Mechanism of the calcium-regulation of muscle contraction--in pursuit of its structural basis. Proceedings of the Japan Academy. Series B, Physical and biological sciences. 2015:91(7):321-50. doi: 10.2183/pjab.91.321. Epub
[PubMed PMID: 26194856]
[15]
Munakomi S, Rajbanshi S, Adhikary PS. Case Report: A giant but silent adrenal pheochromocytoma - a rare entity. F1000Research. 2016:5():290
[PubMed PMID: 27785358]
Level 3 (low-level) evidence
[16]
Cajipe KM, Gonzalez G, Kaushik D. Giant cystic pheochromocytoma. BMJ case reports. 2017 Nov 8:2017():. pii: bcr-2017-222264. doi: 10.1136/bcr-2017-222264. Epub 2017 Nov 8
[PubMed PMID: 29122903]
Level 3 (low-level) evidence
[17]
Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel-Lindau disease. Lancet (London, England). 2003 Jun 14:361(9374):2059-67
[PubMed PMID: 12814730]
[18]
Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, Crossey PA, Webster AR, Affara NA, Ferguson-Smith MA, Brauch H, Glavac D, Neumann HP, Tisherman S, Mulvihill JJ, Gross DJ, Shuin T, Whaley J, Seizinger B, Kley N, Olschwang S, Boisson C, Richard S, Lips CH, Lerman M. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Human mutation. 1996:8(4):348-57
[PubMed PMID: 8956040]
[19]
Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, Mannelli M, Goldstein DS, Linehan WM, Lenders JW, Pacak K. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. The Journal of clinical endocrinology and metabolism. 2001 May:86(5):1999-2008
[PubMed PMID: 11344198]
[20]
Frank-Raue K, Rondot S, Raue F. Molecular genetics and phenomics of RET mutations: Impact on prognosis of MTC. Molecular and cellular endocrinology. 2010 Jun 30:322(1-2):2-7. doi: 10.1016/j.mce.2010.01.012. Epub 2010 Jan 18
[PubMed PMID: 20083156]
[21]
Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nature reviews. Disease primers. 2017 Feb 23:3():17004. doi: 10.1038/nrdp.2017.4. Epub 2017 Feb 23
[PubMed PMID: 28230061]
[22]
DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000 Mar:105(3 Pt 1):608-14
[PubMed PMID: 10699117]
[23]
Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. von Recklinghausen's disease and pheochromocytomas. The Journal of urology. 1999 Nov:162(5):1582-6
[PubMed PMID: 10524872]
[24]
Mannelli M, Lenders JW, Pacak K, Parenti G, Eisenhofer G. Subclinical phaeochromocytoma. Best practice & research. Clinical endocrinology & metabolism. 2012 Aug:26(4):507-15. doi: 10.1016/j.beem.2011.10.008. Epub 2012 May 22
[PubMed PMID: 22863392]
[25]
Kuhweide R, Lanser MJ, Fisch U. Catecholamine-secreting paragangliomas at the skull base. Skull base surgery. 1996:6(1):35-45
[PubMed PMID: 17170951]
[26]
van Duinen N, Steenvoorden D, Kema IP, Jansen JC, Vriends AH, Bayley JP, Smit JW, Romijn JA, Corssmit EP. Increased urinary excretion of 3-methoxytyramine in patients with head and neck paragangliomas. The Journal of clinical endocrinology and metabolism. 2010 Jan:95(1):209-14. doi: 10.1210/jc.2009-1632. Epub 2009 Nov 6
[PubMed PMID: 19897674]
[27]
Pellitteri PK, Rinaldo A, Myssiorek D, Gary Jackson C, Bradley PJ, Devaney KO, Shaha AR, Netterville JL, Manni JJ, Ferlito A. Paragangliomas of the head and neck. Oral oncology. 2004 Jul:40(6):563-75
[PubMed PMID: 15063383]
[28]
Papaspyrou K, Mewes T, Rossmann H, Fottner C, Schneider-Raetzke B, Bartsch O, Schreckenberger M, Lackner KJ, Amedee RG, Mann WJ. Head and neck paragangliomas: Report of 175 patients (1989-2010). Head & neck. 2012 May:34(5):632-7. doi: 10.1002/hed.21790. Epub 2011 Jun 20
[PubMed PMID: 21692132]
[29]
Timmers HJ, Kozupa A, Eisenhofer G, Raygada M, Adams KT, Solis D, Lenders JW, Pacak K. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. The Journal of clinical endocrinology and metabolism. 2007 Mar:92(3):779-86
[PubMed PMID: 17200167]
[30]
Tsujimoto G, Manger WM, Hoffman BB. Desensitization of beta-adrenergic receptors by pheochromocytoma. Endocrinology. 1984 Apr:114(4):1272-8
[PubMed PMID: 6323140]
[31]
Streeten DH, Anderson GH Jr. Mechanisms of orthostatic hypotension and tachycardia in patients with pheochromocytoma. American journal of hypertension. 1996 Aug:9(8):760-9
[PubMed PMID: 8862222]
[32]
Song G, Joe BN, Yeh BM, Meng MV, Westphalen AC, Coakley FV. Risk of catecholamine crisis in patients undergoing resection of unsuspected pheochromocytoma. International braz j urol : official journal of the Brazilian Society of Urology. 2011 Jan-Feb:37(1):35-40;discussion 40-1
[PubMed PMID: 21385478]
[33]
Shen SJ, Cheng HM, Chiu AW, Chou CW, Chen JY. Perioperative hypertensive crisis in clinically silent pheochromocytomas: report of four cases. Chang Gung medical journal. 2005 Jan:28(1):44-50
[PubMed PMID: 15804148]
Level 3 (low-level) evidence
[34]
Kloos RT, Gross MD, Francis IR, Korobkin M, Shapiro B. Incidentally discovered adrenal masses. Endocrine reviews. 1995 Aug:16(4):460-84
[PubMed PMID: 8521790]
[35]
Glazer HS, Weyman PJ, Sagel SS, Levitt RG, McClennan BL. Nonfunctioning adrenal masses: incidental discovery on computed tomography. AJR. American journal of roentgenology. 1982 Jul:139(1):81-5
[PubMed PMID: 6979870]
[36]
Mantero F, Terzolo M, Arnaldi G, Osella G, Masini AM, Alì A, Giovagnetti M, Opocher G, Angeli A. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. The Journal of clinical endocrinology and metabolism. 2000 Feb:85(2):637-44
[PubMed PMID: 10690869]
Level 3 (low-level) evidence
[37]
Lenders JW, Willemsen JJ, Eisenhofer G, Ross HA, Pacak K, Timmers HJ, Sweep CG. Is supine rest necessary before blood sampling for plasma metanephrines? Clinical chemistry. 2007 Feb:53(2):352-4
[PubMed PMID: 17200132]
[38]
de Jong WH, Eisenhofer G, Post WJ, Muskiet FA, de Vries EG, Kema IP. Dietary influences on plasma and urinary metanephrines: implications for diagnosis of catecholamine-producing tumors. The Journal of clinical endocrinology and metabolism. 2009 Aug:94(8):2841-9. doi: 10.1210/jc.2009-0303. Epub 2009 Jun 30
[PubMed PMID: 19567530]
[39]
Deutschbein T, Unger N, Jaeger A, Broecker-Preuss M, Mann K, Petersenn S. Influence of various confounding variables and storage conditions on metanephrine and normetanephrine levels in plasma. Clinical endocrinology. 2010 Aug:73(2):153-60. doi: 10.1111/j.1365-2265.2009.03761.x. Epub 2009 Dec 18
[PubMed PMID: 20039892]
[40]
Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peçzkowska M, Szmigielski C, Eng C, Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. The New England journal of medicine. 2002 May 9:346(19):1459-66
[PubMed PMID: 12000816]
[41]
Sarkadi B, Saskoi E, Butz H, Patocs A. Genetics of Pheochromocytomas and Paragangliomas Determine the Therapeutical Approach. International journal of molecular sciences. 2022 Jan 27:23(3):. doi: 10.3390/ijms23031450. Epub 2022 Jan 27
[PubMed PMID: 35163370]
[42]
Lee JE, Curley SA, Gagel RF, Evans DB, Hickey RC. Cortical-sparing adrenalectomy for patients with bilateral pheochromocytoma. Surgery. 1996 Dec:120(6):1064-70; discussion 1070-1
[PubMed PMID: 8957496]
[43]
Neumann HP, Reincke M, Bender BU, Elsner R, Janetschek G. Preserved adrenocortical function after laparoscopic bilateral adrenal sparing surgery for hereditary pheochromocytoma. The Journal of clinical endocrinology and metabolism. 1999 Aug:84(8):2608-10
[PubMed PMID: 10443647]
[44]
Neumann HPH, Tsoy U, Bancos I, Amodru V, Walz MK, Tirosh A, Kaur RJ, McKenzie T, Qi X, Bandgar T, Petrov R, Yukina MY, Roslyakova A, van der Horst-Schrivers ANA, Berends AMA, Hoff AO, Castroneves LA, Ferrara AM, Rizzati S, Mian C, Dvorakova S, Hasse-Lazar K, Kvachenyuk A, Peczkowska M, Loli P, Erenler F, Krauss T, Almeida MQ, Liu L, Zhu F, Recasens M, Wohllk N, Corssmit EPM, Shafigullina Z, Calissendorff J, Grozinsky-Glasberg S, Kunavisarut T, Schalin-Jäntti C, Castinetti F, Vlcek P, Beltsevich D, Egorov VI, Schiavi F, Links TP, Lechan RM, Bausch B, Young WF Jr, Eng C, International Bilateral-Pheochromocytoma-Registry Group. Comparison of Pheochromocytoma-Specific Morbidity and Mortality Among Adults With Bilateral Pheochromocytomas Undergoing Total Adrenalectomy vs Cortical-Sparing Adrenalectomy. JAMA network open. 2019 Aug 2:2(8):e198898. doi: 10.1001/jamanetworkopen.2019.8898. Epub 2019 Aug 2
[PubMed PMID: 31397861]
Level 2 (mid-level) evidence
[45]
Ulchaker JC, Goldfarb DA, Bravo EL, Novick AC. Successful outcomes in pheochromocytoma surgery in the modern era. The Journal of urology. 1999 Mar:161(3):764-7
[PubMed PMID: 10022680]
[46]
Jimenez C. Treatment for Patients With Malignant Pheochromocytomas and Paragangliomas: A Perspective From the Hallmarks of Cancer. Frontiers in endocrinology. 2018:9():277. doi: 10.3389/fendo.2018.00277. Epub 2018 May 28
[PubMed PMID: 29892268]
Level 3 (low-level) evidence
[47]
Roman-Gonzalez A, Zhou S, Ayala-Ramirez M, Shen C, Waguespack SG, Habra MA, Karam JA, Perrier N, Wood CG, Jimenez C. Impact of Surgical Resection of the Primary Tumor on Overall Survival in Patients With Metastatic Pheochromocytoma or Sympathetic Paraganglioma. Annals of surgery. 2018 Jul:268(1):172-178. doi: 10.1097/SLA.0000000000002195. Epub
[PubMed PMID: 28257320]
[48]
Breen W, Bancos I, Young WF Jr, Bible KC, Laack NN, Foote RL, Hallemeier CL. External beam radiation therapy for advanced/unresectable malignant paraganglioma and pheochromocytoma. Advances in radiation oncology. 2018 Jan-Mar:3(1):25-29. doi: 10.1016/j.adro.2017.11.002. Epub 2017 Nov 22
[PubMed PMID: 29556576]
Level 3 (low-level) evidence
[49]
Fishbein L, Bonner L, Torigian DA, Nathanson KL, Cohen DL, Pryma D, Cengel KA. External beam radiation therapy (EBRT) for patients with malignant pheochromocytoma and non-head and -neck paraganglioma: combination with 131I-MIBG. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2012 May:44(5):405-10. doi: 10.1055/s-0032-1308992. Epub 2012 May 7
[PubMed PMID: 22566196]
[50]
Ayala-Ramirez M, Feng L, Habra MA, Rich T, Dickson PV, Perrier N, Phan A, Waguespack S, Patel S, Jimenez C. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer. 2012 Jun 1:118(11):2804-12. doi: 10.1002/cncr.26577. Epub 2011 Oct 17
[PubMed PMID: 22006217]
[51]
Tanabe A, Naruse M, Nomura K, Tsuiki M, Tsumagari A, Ichihara A. Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine in patients with malignant pheochromocytoma and paraganglioma. Hormones & cancer. 2013 Apr:4(2):103-10. doi: 10.1007/s12672-013-0133-2. Epub 2013 Jan 30
[PubMed PMID: 23361939]
[52]
Niemeijer ND, Alblas G, van Hulsteijn LT, Dekkers OM, Corssmit EP. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis. Clinical endocrinology. 2014 Nov:81(5):642-51. doi: 10.1111/cen.12542. Epub 2014 Jul 30
[PubMed PMID: 25041164]
Level 1 (high-level) evidence
[53]
Huang H, Abraham J, Hung E, Averbuch S, Merino M, Steinberg SM, Pacak K, Fojo T. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer. 2008 Oct 15:113(8):2020-8. doi: 10.1002/cncr.23812. Epub
[PubMed PMID: 18780317]
[54]
van der Harst E, de Herder WW, Bruining HA, Bonjer HJ, de Krijger RR, Lamberts SW, van de Meiracker AH, Boomsma F, Stijnen T, Krenning EP, Bosman FT, Kwekkeboom DJ. [(123)I]metaiodobenzylguanidine and [(111)In]octreotide uptake in begnign and malignant pheochromocytomas. The Journal of clinical endocrinology and metabolism. 2001 Feb:86(2):685-93
[PubMed PMID: 11158032]
[55]
Loh KC, Fitzgerald PA, Matthay KK, Yeo PP, Price DC. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): a comprehensive review of 116 reported patients. Journal of endocrinological investigation. 1997 Dec:20(11):648-58
[PubMed PMID: 9492103]
[56]
Mukherjee JJ, Kaltsas GA, Islam N, Plowman PN, Foley R, Hikmat J, Britton KE, Jenkins PJ, Chew SL, Monson JP, Besser GM, Grossman AB. Treatment of metastatic carcinoid tumours, phaeochromocytoma, paraganglioma and medullary carcinoma of the thyroid with (131)I-meta-iodobenzylguanidine [(131)I-mIBG]. Clinical endocrinology. 2001 Jul:55(1):47-60
[PubMed PMID: 11453952]
[57]
Troncone L, Rufini V. Nuclear medicine therapy of pheochromocytoma and paraganglioma. The quarterly journal of nuclear medicine : official publication of the Italian Association of Nuclear Medicine (AIMN) [and] the International Association of Radiopharmacology (IAR). 1999 Dec:43(4):344-55
[PubMed PMID: 10731785]
[58]
Sisson JC. Radiopharmaceutical treatment of pheochromocytomas. Annals of the New York Academy of Sciences. 2002 Sep:970():54-60
[PubMed PMID: 12381541]
[59]
Hao Z, Sadek I. Sunitinib: the antiangiogenic effects and beyond. OncoTargets and therapy. 2016:9():5495-505. doi: 10.2147/OTT.S112242. Epub 2016 Sep 8
[PubMed PMID: 27660467]
[60]
O'Kane GM, Ezzat S, Joshua AM, Bourdeau I, Leibowitz-Amit R, Olney HJ, Krzyzanowska M, Reuther D, Chin S, Wang L, Brooks K, Hansen AR, Asa SL, Knox JJ. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: the SNIPP trial. British journal of cancer. 2019 Jun:120(12):1113-1119. doi: 10.1038/s41416-019-0474-x. Epub 2019 May 20
[PubMed PMID: 31105270]
[61]
Ayala-Ramirez M, Chougnet CN, Habra MA, Palmer JL, Leboulleux S, Cabanillas ME, Caramella C, Anderson P, Al Ghuzlan A, Waguespack SG, Deandreis D, Baudin E, Jimenez C. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. The Journal of clinical endocrinology and metabolism. 2012 Nov:97(11):4040-50. doi: 10.1210/jc.2012-2356. Epub 2012 Sep 10
[PubMed PMID: 22965939]
[62]
Amar L, Servais A, Gimenez-Roqueplo AP, Zinzindohoue F, Chatellier G, Plouin PF. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. The Journal of clinical endocrinology and metabolism. 2005 Apr:90(4):2110-6
[PubMed PMID: 15644401]
[63]
Holscher I, van den Berg TJ, Dreijerink KMA, Engelsman AF, Nieveen van Dijkum EJM. Recurrence Rate of Sporadic Pheochromocytomas After Curative Adrenalectomy: A Systematic Review and Meta-analysis. The Journal of clinical endocrinology and metabolism. 2021 Jan 23:106(2):588-597. doi: 10.1210/clinem/dgaa794. Epub
[PubMed PMID: 33125073]
Level 1 (high-level) evidence
[64]
Harrington JL, Farley DR, van Heerden JA, Ramin KD. Adrenal tumors and pregnancy. World journal of surgery. 1999 Feb:23(2):182-6
[PubMed PMID: 9880429]
[65]
Lenders JWM, Langton K, Langenhuijsen JF, Eisenhofer G. Pheochromocytoma and Pregnancy. Endocrinology and metabolism clinics of North America. 2019 Sep:48(3):605-617. doi: 10.1016/j.ecl.2019.05.006. Epub 2019 Jun 13
[PubMed PMID: 31345526]
[66]
Bancos I, Atkinson E, Eng C, Young WF Jr, Neumann HPH, International Pheochromocytoma and Pregnancy Study Group. Maternal and fetal outcomes in phaeochromocytoma and pregnancy: a multicentre retrospective cohort study and systematic review of literature. The lancet. Diabetes & endocrinology. 2021 Jan:9(1):13-21. doi: 10.1016/S2213-8587(20)30363-6. Epub 2020 Nov 26
[PubMed PMID: 33248478]
Level 2 (mid-level) evidence
[67]
van der Weerd K, van Noord C, Loeve M, Knapen MFCM, Visser W, de Herder WW, Franssen G, van der Marel CD, Feelders RA. ENDOCRINOLOGY IN PREGNANCY: Pheochromocytoma in pregnancy: case series and review of literature. European journal of endocrinology. 2017 Aug:177(2):R49-R58. doi: 10.1530/EJE-16-0920. Epub 2017 Apr 5
[PubMed PMID: 28381449]
Level 2 (mid-level) evidence