Learning Outcome
- Describe the presentation of sickle cell anemia
- List the complications of sickle cell anemia
- Summarize the management of sickle cell anemia
- Recall the sickle cell crisis

Sickle cell anemia is the most severe form of sickle cell disease and is the homozygous state for hemoglobin S. Sickle cell anemia is prevalent in Africa, the Middle East, and parts of India. It is common in geographical areas where malaria is widespread. Hemoglobin in most individuals is present in soluble form. However, in sickle cell disease, hemoglobin precipitates as insoluble crystals, leading to an abnormal shape and size of RBCs and subsequent phagocytosis of the affected corpuscles.[1][2][3]
A point mutation in the beta-globin chain of the hemoglobin causes sickle cell disease. Specifically, it occurs when a single base from A to T in the codon for glutamic acid at position 6 is changed to valine of the beta-globin. If this mutation affects both beta-globin chains, sickle cell anemia occurs; if only one chain is involved, it results in the sickle cell trait.[4]
This disease is predominantly present in individuals of African origin but also affects people of Middle Eastern, Indian, and Mediterranean descent. It is estimated that 1 in 13 children of African descent suffers from the sickle cell trait. Sickle cell disease affects 1 in 365 individuals of African descent; in America, about 100,000 individuals currently suffer from this disease.[5]
Sickle cell disease usually manifests after six months of age when fetal hemoglobin levels begin to fall. This timing occurs because fetal hemoglobin keeps the sickle cell hemoglobin in solubilized form. The most common presenting feature is vaso-occlusive crises. These vaso-occlusive crises may present in a variety of ways. Patients commonly complain of excruciating pain in the abdomen, thorax, joints, long bones, and digits. Some individuals may experience multiple episodes, while others may remain free of them for long periods of time. Signs and symptoms of anemia are also prevalent, including palpitations, fatigue, pallor, and tachycardia. Repeated vaso-occlusive crises may result in splenic infarctions and resultant functional asplenia. This asplenia results in repeated infections with encapsulated bacteria like Streptococcus pneumoniae, Staphylococcus aureus, and many others. These pathogens may cause life-threatening pneumonia and septicemia, which are usually fatal.
Vaso-occlusive crises of the digits present as dactylitis, where the finger becomes severely painful, red, hot, and swollen. Abdominal vaso-occlusion usually mimics the acute abdomen in pain severity. Acute chest syndrome may present as chest pain, cough, leukocytosis, tachypnea, and respiratory difficulty in young children. These may be fatal. Sickle cell anemia patients may also present with splenic sequestration crisis. This condition occurs when sickle-shaped cells get entangled in the splenic pulp, leading to severe anemia with a rapidly enlarging spleen. A stroke is the most critical central nervous system condition caused by sickle cell disease. Retinal hemorrhages with visual loss are also critical conditions.
Repeated cycles of hemolysis lead to increased pigment load. This increase results in pigmented stone formation in the gallbladder, inciting cholelithiases. Sickling in the renal vasculature causes isosthenuria, a condition where the kidneys lose their ability to concentrate the urine appropriately. It also occludes the penile vasculature, which causes a prolonged, painful erection known as priapism. Avascular necrosis of the long bones, particularly the head of the femur, is also a troublesome condition. It involves the slow and gradual destruction of the articular surface of the femoral head, which requires hemiarthroplasty to restore mobility.
Aplastic crises are another significant manifestation of sickle cell disease. Here, when the presence of parvovirus B19 challenges an already stressed bone marrow, it fails to generate the appropriate number of RBCs which results in severe anemia.
Newborns with a family positive for sickle cell disease should undergo a screening test. This screening test for sickle cell hemoglobin is mandatory in the United States. Prenatal diagnosis in genetically prone fetuses can be made using the chorionic villus sampling technique or amniocentesis.[6]
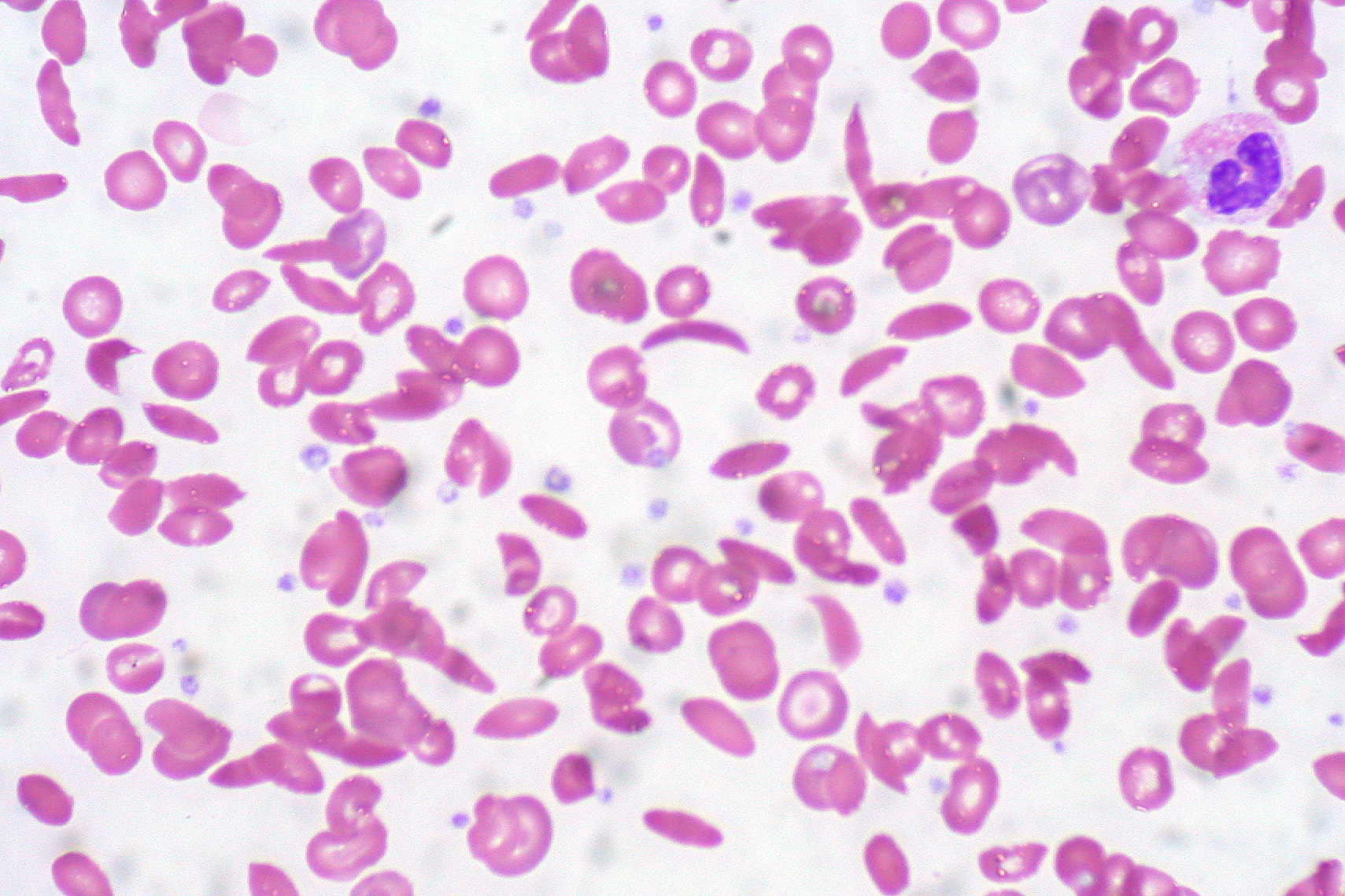
A complete blood count with a peripheral picture is the initial test. Here, a reduced RBC number, reduced reticulocyte count, variable mean corpuscular volume, increased leukocyte count, reduced ESR, and the presence of sickle-shaped cells in the periphery usually indicate the diagnosis of sickle cell disease. The presence of Howell-Jolly bodies indicates functional asplenia in the patient. Subsequent hemoglobin electrophoresis confirms the diagnosis of sickle cell disease if the concentration of sickle cell hemoglobin is more than 90% of the total hemoglobin and the fetal hemoglobin comprises the rest of the hemoglobin isotype. However, if the concentration of sickle cell hemoglobin is at or around 45%, it indicates the presence of the sickle cell trait rather than the disease itself.
A urine analysis should be performed to rule out a urinary tract infection as a cause of hematuria and identify isosthenuria in sickle cell disease patients.
If a patient presents to the emergency department with an acute vaso-occlusive episode and the diagnosis of sickle cell disease is not yet established, one can administer an instant sickling test which may test positive for sickle cell hemoglobin. However, this test has a limiting factor. It can not differentiate between heterozygous and homozygous states of sickle cell hemoglobin.
Arterial blood gases are required to monitor pulmonary functions in case of acute chest syndrome. Serial arterial blood gases shall be obtained to monitor the severity of the pulmonary crises.
A chest X-ray shall be performed in patients with respiratory signs and symptoms, but it may be normal in the early phase of acute chest syndrome. Plain radiography of the peripheries is used in identifying acute and subacute marrow infarctions, as well as observing old infarcts. In the case of osteomyelitis, early radiographs are not useful because they do not show significant changes. However, in the subsequent two weeks, the plain radiographs may reveal the destruction of the bone, periosteal bone formation, sinus tract, and sequestra.
MRI scans are paramount in diagnosing osteomyelitis and avascular necrosis of the femoral and humeral heads. Technetium-99 scans are also used to detect osteonecrosis.
The treatment of sickle cell disease has seven major goals:
Pharmacotherapy of sickle cell disease usually revolves around preventing its complications. Hydroxyurea is an antimetabolite known to increase the fetal hemoglobin levels in the circulation of RBCs; fetal hemoglobin keeps hemoglobin in soluble form and prevents polymerization of the sickle cell hemoglobin, thus preventing most complications of sickle cell disease.[7][8][9]
Opioids, NSAIDs, steroids, antiemetics, tricyclic antidepressants, and antibiotics are all used to cure or ameliorate the complications of the disease.
Vaccines, particularly against encapsulated bacteria, help prevent life-threatening infections as sickle cell disease patients are usually functionally asplenic. Folic acid supplementation prevents macrocytic anemia.
A bone marrow transplant is curative in sickle cell disease patients.
A blood transfusion, particularly in aplastic crises, is also essential.
Sickle cell anemia is a severe genetic disorder with high morbidity and mortality. The disease usually manifests early in life and can present with several types of occlusive crises.
Screening for sickle cell anemia is mandatory at birth in the United States, allowing for early diagnosis and treatment. Because the disorder affects almost every organ system in the body, an interprofessional approach is necessary to ensure adequate treatment and prevent complications. However, as the population ages, chronic complications like pulmonary hypertension are emerging with very high morbidity and mortality. The consensus among experts is that sickle cell should be managed by an interprofessional group of healthcare professionals, including physical therapists, psychiatrists, social workers, nurses, pharmacists, substance abuse counselors, pain counselors, and rehabilitation specialists.[10][11] [Level 5] Anytime there is a fever, an infectious disease consult should be made promptly.
An orthopedic surgeon should be consulted for hip pain or difficulty with gait. A radiologist is essential for obtaining samples from bone if osteomyelitis is suspected. These patients need thorough eye exams by an ophthalmologist, and a urologist is needed to manage priapism.
Most of these patients are on many medications. The pharmacist plays a critical role in ensuring the patient remains compliant with medications and free from adverse drug effects.
The nurse should educate the patient on the importance of remaining hydrated, getting the right vaccinations, and ensuring follow-up with the respective healthcare providers.
Outcomes
The outcome for most sickle cell patients is mixed. The goal is to achieve an average lifespan with minimal morbidity and mortality. However, many of these individuals die prematurely despite improvements in treatment. The morbidity is very high, and nearly all individuals experience a vaso-occlusive crisis at some point in their lives. These patients often are not able to work due to their disability and live a poor quality of life with chronic pain. The leading causes of death are acute chest syndrome, pulmonary embolism, and infection. Outside of North America, the life expectancy of sickle cell patients is in the 30s and 40s. Many guidelines have been developed to manage sickle cell disease. They include penicillin prophylaxis for children, blood transfusions, and pneumococcal vaccination. The drug hydroxyurea has made it possible for patients to live longer than ever.[12] [Level 5]
Call a clinician if:
The key to improved outcomes is patient education. The earlier one seeks medical help, the better the outcomes, and thus patients should seek help in the presence of:
At the same time, patients should avoid the following:
Patients should be urged to:
The patient should be educated about hydroxyurea since the evidence shows that the drug can reduce vaso-occlusive crises.
Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet (London, England). 2010 Dec 11:376(9757):2018-31. doi: 10.1016/S0140-6736(10)61029-X. Epub 2010 Dec 3 [PubMed PMID: 21131035]
Eaton WA. Linus Pauling and sickle cell disease. Biophysical chemistry. 2003:100(1-3):109-16 [PubMed PMID: 12646356]
Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, Smith WR, Panepinto JA, Weatherall DJ, Costa FF, Vichinsky EP. Sickle cell disease. Nature reviews. Disease primers. 2018 Mar 15:4():18010. doi: 10.1038/nrdp.2018.10. Epub 2018 Mar 15 [PubMed PMID: 29542687]
Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, Nathan DG. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. The Journal of clinical investigation. 1984 Aug:74(2):652-6 [PubMed PMID: 6205021]
Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. American journal of hematology. 2012 Aug:87(8):795-803. doi: 10.1002/ajh.23232. Epub 2012 May 28 [PubMed PMID: 22641398]
Naoum PC. Sickle cell disease: from the beginning until it was recognized as a public health disease. Revista brasileira de hematologia e hemoterapia. 2011:33(1):7-9. doi: 10.5581/1516-8484.20110006. Epub [PubMed PMID: 23284235]
Lubeck D, Agodoa I, Bhakta N, Danese M, Pappu K, Howard R, Gleeson M, Halperin M, Lanzkron S. Estimated Life Expectancy and Income of Patients With Sickle Cell Disease Compared With Those Without Sickle Cell Disease. JAMA network open. 2019 Nov 1:2(11):e1915374. doi: 10.1001/jamanetworkopen.2019.15374. Epub 2019 Nov 1 [PubMed PMID: 31730182]
Pecker LH, Schaefer BA, Luchtman-Jones L. Knowledge insufficient: the management of haemoglobin SC disease. British journal of haematology. 2017 Feb:176(4):515-526. doi: 10.1111/bjh.14444. Epub 2016 Dec 16 [PubMed PMID: 27982424]
Mangla A, Hamad H. Pure Red Cell Aplasia. StatPearls. 2024 Jan:(): [PubMed PMID: 31751023]
Remacha A, Sanz C, Contreras E, De Heredia CD, Grifols JR, Lozano M, Nuñez GM, Salinas R, Corral M, Villegas A, Spanish Society of Blood Transfusion, Spanish Society of Haematology and Haemotherapy. Guidelines on haemovigilance of post-transfusional iron overload. Blood transfusion = Trasfusione del sangue. 2013 Jan:11(1):128-39. doi: 10.2450/2012.0114-11. Epub 2012 Jul 4 [PubMed PMID: 22790272]
Brittenham GM, Griffith PM, Nienhuis AW, McLaren CE, Young NS, Tucker EE, Allen CJ, Farrell DE, Harris JW. Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major. The New England journal of medicine. 1994 Sep 1:331(9):567-73 [PubMed PMID: 8047080]
Jenerette CM, Brewer C. Health-related stigma in young adults with sickle cell disease. Journal of the National Medical Association. 2010 Nov:102(11):1050-5 [PubMed PMID: 21141294]
Kato GJ. Priapism in sickle-cell disease: a hematologist's perspective. The journal of sexual medicine. 2012 Jan:9(1):70-8. doi: 10.1111/j.1743-6109.2011.02287.x. Epub 2011 May 6 [PubMed PMID: 21554552]
Howard J. Sickle cell disease: when and how to transfuse. Hematology. American Society of Hematology. Education Program. 2016 Dec 2:2016(1):625-631 [PubMed PMID: 27913538]
Thom CS, Dickson CF, Gell DA, Weiss MJ. Hemoglobin variants: biochemical properties and clinical correlates. Cold Spring Harbor perspectives in medicine. 2013 Mar 1:3(3):a011858. doi: 10.1101/cshperspect.a011858. Epub 2013 Mar 1 [PubMed PMID: 23388674]
Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP, Bonds DR. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. The New England journal of medicine. 1995 May 18:332(20):1317-22 [PubMed PMID: 7715639]
Wang WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, Rana S, Thornburg CD, Rogers ZR, Kalpatthi RV, Barredo JC, Brown RC, Sarnaik SA, Howard TH, Wynn LW, Kutlar A, Armstrong FD, Files BA, Goldsmith JC, Waclawiw MA, Huang X, Thompson BW, BABY HUG investigators. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet (London, England). 2011 May 14:377(9778):1663-72. doi: 10.1016/S0140-6736(11)60355-3. Epub [PubMed PMID: 21571150]
Thompson A. A Targeted Agent for Sickle Cell Disease - Changing the Protein but Not the Gene. The New England journal of medicine. 2019 Aug 8:381(6):579-580. doi: 10.1056/NEJMe1906771. Epub 2019 Jun 14 [PubMed PMID: 31199089]
Vichinsky E, Hoppe CC, Ataga KI, Ware RE, Nduba V, El-Beshlawy A, Hassab H, Achebe MM, Alkindi S, Brown RC, Diuguid DL, Telfer P, Tsitsikas DA, Elghandour A, Gordeuk VR, Kanter J, Abboud MR, Lehrer-Graiwer J, Tonda M, Intondi A, Tong B, Howard J, HOPE Trial Investigators. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. The New England journal of medicine. 2019 Aug 8:381(6):509-519. doi: 10.1056/NEJMoa1903212. Epub 2019 Jun 14 [PubMed PMID: 31199090]
Blair HA. Crizanlizumab: First Approval. Drugs. 2020 Jan:80(1):79-84. doi: 10.1007/s40265-019-01254-2. Epub [PubMed PMID: 31933169]
Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, Guthrie TH, Knight-Madden J, Alvarez OA, Gordeuk VR, Gualandro S, Colella MP, Smith WR, Rollins SA, Stocker JW, Rother RP. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. The New England journal of medicine. 2017 Feb 2:376(5):429-439. doi: 10.1056/NEJMoa1611770. Epub 2016 Dec 3 [PubMed PMID: 27959701]
Sadaf A, Quinn CT. L-glutamine for sickle cell disease: Knight or pawn? Experimental biology and medicine (Maywood, N.J.). 2020 Jan:245(2):146-154. doi: 10.1177/1535370219900637. Epub 2020 Jan 27 [PubMed PMID: 31985279]
Niihara Y, Miller ST, Kanter J, Lanzkron S, Smith WR, Hsu LL, Gordeuk VR, Viswanathan K, Sarnaik S, Osunkwo I, Guillaume E, Sadanandan S, Sieger L, Lasky JL, Panosyan EH, Blake OA, New TN, Bellevue R, Tran LT, Razon RL, Stark CW, Neumayr LD, Vichinsky EP, Investigators of the Phase 3 Trial of l-Glutamine in Sickle Cell Disease. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. The New England journal of medicine. 2018 Jul 19:379(3):226-235. doi: 10.1056/NEJMoa1715971. Epub [PubMed PMID: 30021096]
de la Fuente J, Gluckman E, Makani J, Telfer P, Faulkner L, Corbacioglu S, Paediatric Diseases Working Party of the European Society for Blood and Marrow Transplantation. The role of haematopoietic stem cell transplantation for sickle cell disease in the era of targeted disease-modifying therapies and gene editing. The Lancet. Haematology. 2020 Dec:7(12):e902-e911. doi: 10.1016/S2352-3026(20)30283-0. Epub [PubMed PMID: 33242447]
Robinson TM, Fuchs EJ. Allogeneic stem cell transplantation for sickle cell disease. Current opinion in hematology. 2016 Nov:23(6):524-529 [PubMed PMID: 27496639]
Esrick EB, Lehmann LE, Biffi A, Achebe M, Brendel C, Ciuculescu MF, Daley H, MacKinnon B, Morris E, Federico A, Abriss D, Boardman K, Khelladi R, Shaw K, Negre H, Negre O, Nikiforow S, Ritz J, Pai SY, London WB, Dansereau C, Heeney MM, Armant M, Manis JP, Williams DA. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. The New England journal of medicine. 2021 Jan 21:384(3):205-215. doi: 10.1056/NEJMoa2029392. Epub 2020 Dec 5 [PubMed PMID: 33283990]
Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, Foell J, de la Fuente J, Grupp S, Handgretinger R, Ho TW, Kattamis A, Kernytsky A, Lekstrom-Himes J, Li AM, Locatelli F, Mapara MY, de Montalembert M, Rondelli D, Sharma A, Sheth S, Soni S, Steinberg MH, Wall D, Yen A, Corbacioglu S. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. The New England journal of medicine. 2021 Jan 21:384(3):252-260. doi: 10.1056/NEJMoa2031054. Epub 2020 Dec 5 [PubMed PMID: 33283989]
Broder MS, Quock TP, Chang E, Reddy SR, Agarwal-Hashmi R, Arai S, Villa KF. The Cost of Hematopoietic Stem-Cell Transplantation in the United States. American health & drug benefits. 2017 Oct:10(7):366-374 [PubMed PMID: 29263771]
Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. The New England journal of medicine. 1994 Jun 9:330(23):1639-44 [PubMed PMID: 7993409]
Maitra P, Caughey M, Robinson L, Desai PC, Jones S, Nouraie M, Gladwin MT, Hinderliter A, Cai J, Ataga KI. Risk factors for mortality in adult patients with sickle cell disease: a meta-analysis of studies in North America and Europe. Haematologica. 2017 Apr:102(4):626-636. doi: 10.3324/haematol.2016.153791. Epub 2017 Jan 19 [PubMed PMID: 28104703]
Stanton MV, Jonassaint CR, Bartholomew FB, Edwards C, Richman L, DeCastro L, Williams R. The association of optimism and perceived discrimination with health care utilization in adults with sickle cell disease. Journal of the National Medical Association. 2010 Nov:102(11):1056-63 [PubMed PMID: 21141295]