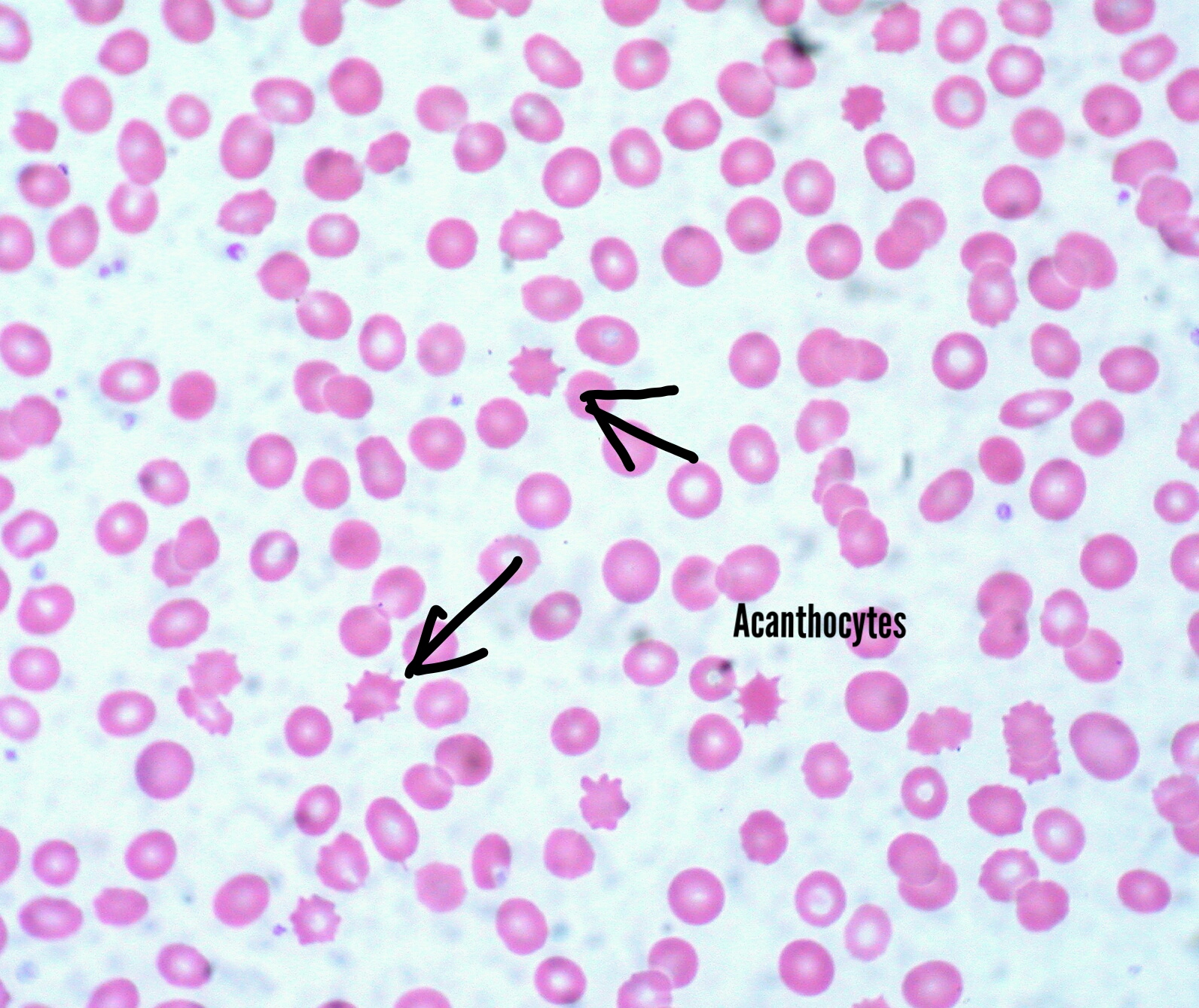
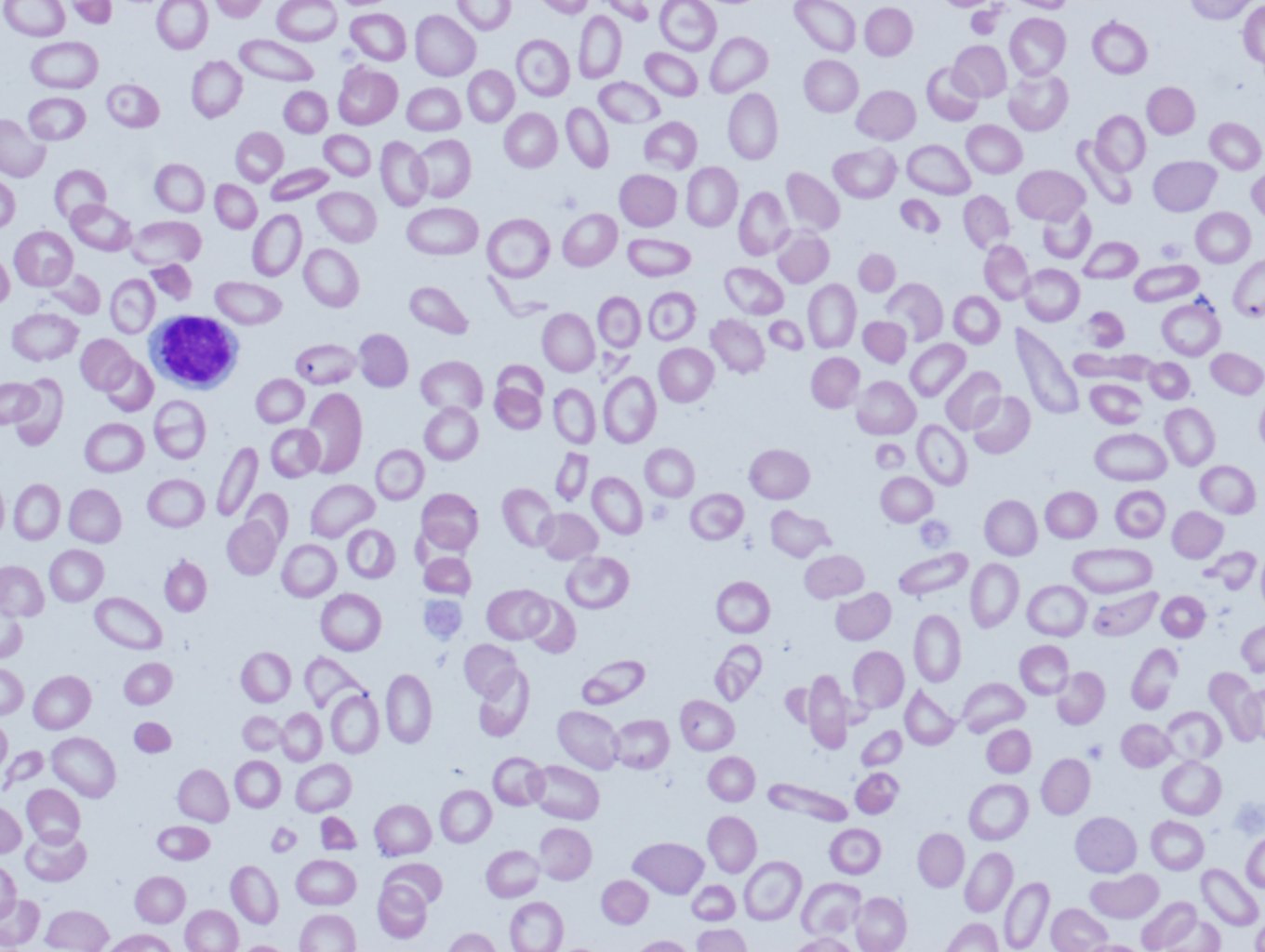
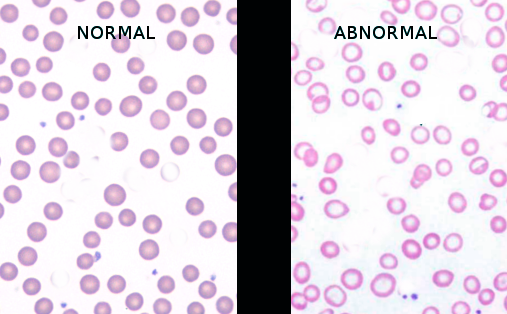
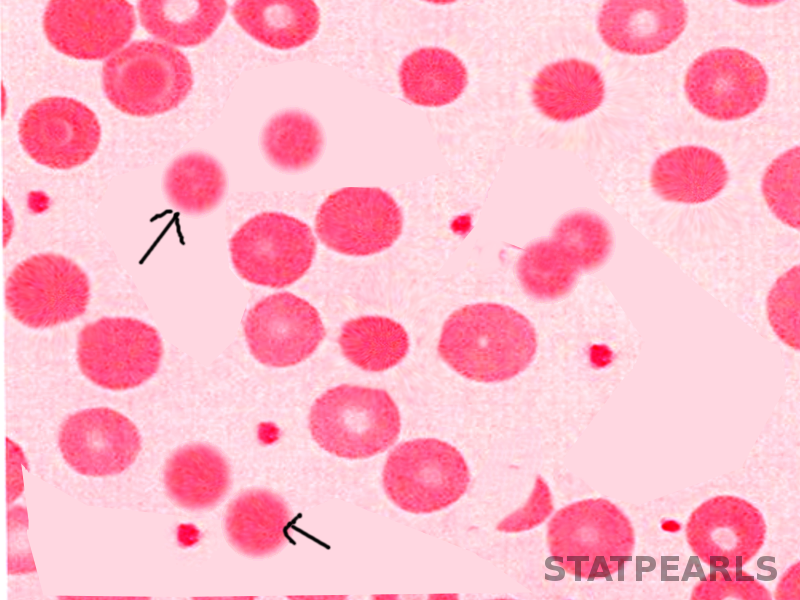
[3]
Narla J, Mohandas N. Red cell membrane disorders. International journal of laboratory hematology. 2017 May:39 Suppl 1():47-52. doi: 10.1111/ijlh.12657. Epub
[PubMed PMID: 28447420]
[5]
Green R, Datta Mitra A. Megaloblastic Anemias: Nutritional and Other Causes. The Medical clinics of North America. 2017 Mar:101(2):297-317. doi: 10.1016/j.mcna.2016.09.013. Epub 2016 Dec 14
[PubMed PMID: 28189172]
[7]
Martin A, Thompson AA. Thalassemias. Pediatric clinics of North America. 2013 Dec:60(6):1383-91. doi: 10.1016/j.pcl.2013.08.008. Epub 2013 Oct 4
[PubMed PMID: 24237977]
[8]
Manciu S, Matei E, Trandafir B. Hereditary Spherocytosis - Diagnosis, Surgical Treatment and Outcomes. A Literature Review. Chirurgia (Bucharest, Romania : 1990). 2017 Mar-Apr:112(2):110-116. doi: 10.21614/chirurgia.112.2.110. Epub
[PubMed PMID: 28463670]
[9]
Adewoyin AS, Nwogoh B. Peripheral blood film - a review. Annals of Ibadan postgraduate medicine. 2014 Dec:12(2):71-9
[PubMed PMID: 25960697]
[12]
Usuki K. [Anemia: From Basic Knowledge to Up-to-Date Treatment. Topic: IV. Hemolytic anemia: Diagnosis and treatment]. Nihon Naika Gakkai zasshi. The Journal of the Japanese Society of Internal Medicine. 2015 Jul 10:104(7):1389-96
[PubMed PMID: 26513958]
[13]
Zini G, d'Onofrio G, Erber WN, Lee SH, Nagai Y, Basak GW, Lesesve JF, International Council for Standardization in Hematology (ICSH). 2021 update of the 2012 ICSH Recommendations for identification, diagnostic value, and quantitation of schistocytes: Impact and revisions. International journal of laboratory hematology. 2021 Dec:43(6):1264-1271. doi: 10.1111/ijlh.13682. Epub 2021 Aug 24
[PubMed PMID: 34431220]
[14]
Desai K, Sridhar A, Matos J, Mulla S, Thirumaran R. Thrombotic Thrombocytopenia Masquerading As COVID-19 Infection. Cureus. 2022 Jul:14(7):e26933. doi: 10.7759/cureus.26933. Epub 2022 Jul 17
[PubMed PMID: 35989804]
[15]
Hasler CR, Owen GR, Brunner W, Reinhart WH. Echinocytosis induced by haemodialysis. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 1998 Dec:13(12):3132-7
[PubMed PMID: 9870478]
[16]
Koralkova P, van Solinge WW, van Wijk R. Rare hereditary red blood cell enzymopathies associated with hemolytic anemia - pathophysiology, clinical aspects, and laboratory diagnosis. International journal of laboratory hematology. 2014 Jun:36(3):388-97. doi: 10.1111/ijlh.12223. Epub
[PubMed PMID: 24750686]
[17]
Mallah HS, Brown MR, Rossi TM, Block RC. Parenteral fish oil-associated burr cell anemia. The Journal of pediatrics. 2010 Feb:156(2):324-6.e1. doi: 10.1016/j.jpeds.2009.07.062. Epub
[PubMed PMID: 20105643]
[18]
Huang S, Zhang J, Tao M, Lv Y, Xu L, Liang Z. Two case reports of chorea-acanthocytosis and review of literature. European journal of medical research. 2022 Feb 7:27(1):22. doi: 10.1186/s40001-022-00646-7. Epub 2022 Feb 7
[PubMed PMID: 35130982]
Level 3 (low-level) evidence
[19]
Peikert K, Hermann A, Danek A. XK-Associated McLeod Syndrome: Nonhematological Manifestations and Relation to VPS13A Disease. Transfusion medicine and hemotherapy : offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und Immunhamatologie. 2022 Feb:49(1):4-12. doi: 10.1159/000521417. Epub 2022 Jan 25
[PubMed PMID: 35221863]
[20]
Yu Y, Lu Y, Wang F, Lu Y, Xie B, Meng X, Tang Y. Acanthocytes Identified in Huntington's Disease. Frontiers in neuroscience. 2022:16():913401. doi: 10.3389/fnins.2022.913401. Epub 2022 Jun 6
[PubMed PMID: 35733931]
[21]
Cloos AS, Daenen LGM, Maja M, Stommen A, Vanderroost J, Van Der Smissen P, Rab M, Westerink J, Mignolet E, Larondelle Y, Terrasi R, Muccioli GG, Dumitru AC, Alsteens D, van Wijk R, Tyteca D. Impaired Cytoskeletal and Membrane Biophysical Properties of Acanthocytes in Hypobetalipoproteinemia - A Case Study. Frontiers in physiology. 2021:12():638027. doi: 10.3389/fphys.2021.638027. Epub 2021 Feb 23
[PubMed PMID: 33708142]
Level 3 (low-level) evidence
[22]
Robier C, Klescher D, Reicht G, Amouzadeh-Ghadikolai O, Quehenberger F, Neubauer M. Dacryocytes are a common morphologic feature of autoimmune and microangiopathic haemolytic anaemia. Clinical chemistry and laboratory medicine. 2015 Jun:53(7):1073-6. doi: 10.1515/cclm-2014-0936. Epub
[PubMed PMID: 25503671]
[23]
Kjelland JD, Dwyre DM, Jonas BA. Acquired Elliptocytosis as a Manifestation of Myelodysplastic Syndrome with Ring Sideroblasts and Multilineage Dysplasia. Case reports in hematology. 2017:2017():3625946. doi: 10.1155/2017/3625946. Epub 2017 Oct 11
[PubMed PMID: 29158926]
Level 3 (low-level) evidence
[24]
Quigley M, Linfesty RL, Bethel K, Sharpe R. Stubby elliptocytes are an invariable feature of leukoerythroblastosis. Blood. 2007 Mar 15:109(6):2666
[PubMed PMID: 17341668]
[25]
Jiménez Gonzalo FJ, de Luis Navarro J, de Blas Orlando JM, Martín Noya A. [Hereditary elliptocytosis associated with heterozygous beta-thalassemia with a hemolytic component]. Sangre. 1999 Oct:44(5):391-2
[PubMed PMID: 10618922]
[26]
Rodgers MS, Chang CC, Kass L. Elliptocytes and tailed poikilocytes correlate with severity of iron-deficiency anemia. American journal of clinical pathology. 1999 May:111(5):672-5
[PubMed PMID: 10230358]
[27]
Kachur ME, Rosen BJ. Educational Case: Anemia in a Neonate. Academic pathology. 2021 Jan-Dec:8():23742895211002829. doi: 10.1177/23742895211002829. Epub 2021 Apr 8
[PubMed PMID: 33889717]
Level 3 (low-level) evidence
[28]
Prchal JT, Gregg XT. Red cell enzymes. Hematology. American Society of Hematology. Education Program. 2005:():19-23
[PubMed PMID: 16304354]
[29]
Lee AC, Aung L, Yip YY, Hia CP. Hereditary stomatocytosis: an unusual cause of severe neonatal jaundice. Singapore medical journal. 2018 Sep:59(9):505. doi: 10.11622/smedj.2018115. Epub
[PubMed PMID: 30310921]
[30]
Flatt JF, Bruce LJ. The Molecular Basis for Altered Cation Permeability in Hereditary Stomatocytic Human Red Blood Cells. Frontiers in physiology. 2018:9():367. doi: 10.3389/fphys.2018.00367. Epub 2018 Apr 16
[PubMed PMID: 29713289]
[31]
Moerdler S, Manwani D. New insights into the pathophysiology and development of novel therapies for sickle cell disease. Hematology. American Society of Hematology. Education Program. 2018 Nov 30:2018(1):493-506. doi: 10.1182/asheducation-2018.1.493. Epub
[PubMed PMID: 30504350]
[32]
Salinas Cisneros G, Thein SL. Research in Sickle Cell Disease: From Bedside to Bench to Bedside. HemaSphere. 2021 Jun:5(6):e584. doi: 10.1097/HS9.0000000000000584. Epub 2021 Jun 1
[PubMed PMID: 34095767]
[33]
Mack AK, Kato GJ. Sickle cell disease and nitric oxide: a paradigm shift? The international journal of biochemistry & cell biology. 2006:38(8):1237-43
[PubMed PMID: 16517208]
[34]
Kavanagh PL, Fasipe TA, Wun T. Sickle Cell Disease: A Review. JAMA. 2022 Jul 5:328(1):57-68. doi: 10.1001/jama.2022.10233. Epub
[PubMed PMID: 35788790]
[35]
Eaton WA, Bunn HF. Treating sickle cell disease by targeting HbS polymerization. Blood. 2017 May 18:129(20):2719-2726. doi: 10.1182/blood-2017-02-765891. Epub 2017 Apr 6
[PubMed PMID: 28385699]
[36]
Wu Y, Liao L, Lin F. The diagnostic protocol for hereditary spherocytosis-2021 update. Journal of clinical laboratory analysis. 2021 Dec:35(12):e24034. doi: 10.1002/jcla.24034. Epub 2021 Oct 24
[PubMed PMID: 34689357]
[37]
Karlsson LK, Mottelson MN, Helby J, Petersen J, Glenthøj A. Acquired spherocytosis in the setting of myelodysplasia. Leukemia research reports. 2022:17():100332. doi: 10.1016/j.lrr.2022.100332. Epub 2022 Jun 2
[PubMed PMID: 35720514]
[39]
Anil More T, Kedar P. Unravelling the genetic and phenotypic heterogeneity of SPTA1 gene variants in Hereditary Elliptocytosis and Hereditary Pyropoikilocytosis patients using next-generation sequencing. Gene. 2022 Nov 15:843():146796. doi: 10.1016/j.gene.2022.146796. Epub 2022 Aug 9
[PubMed PMID: 35961434]
[40]
Bayhan T, Ünal Ş, Gümrük F. Hereditary Elliptocytosis with Pyropoikilocytosis. Turkish journal of haematology : official journal of Turkish Society of Haematology. 2016 Mar 5:33(1):86-7. doi: 10.4274/tjh.2015.0054. Epub 2015 Aug 6
[PubMed PMID: 26377499]
[41]
Opsahl M, Chen W. Blister and bite cells in G6PD deficiency. International journal of laboratory hematology. 2022 Feb:44(1):55-56. doi: 10.1111/ijlh.13689. Epub 2021 Aug 24
[PubMed PMID: 34431226]
[42]
Andolfo I, Alper SL, De Franceschi L, Auriemma C, Russo R, De Falco L, Vallefuoco F, Esposito MR, Vandorpe DH, Shmukler BE, Narayan R, Montanaro D, D'Armiento M, Vetro A, Limongelli I, Zuffardi O, Glader BE, Schrier SL, Brugnara C, Stewart GW, Delaunay J, Iolascon A. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood. 2013 May 9:121(19):3925-35, S1-12. doi: 10.1182/blood-2013-02-482489. Epub 2013 Mar 11
[PubMed PMID: 23479567]
[43]
Gallagher PG. Disorders of red cell volume regulation. Current opinion in hematology. 2013 May:20(3):201-7. doi: 10.1097/MOH.0b013e32835f6870. Epub
[PubMed PMID: 23519154]
Level 3 (low-level) evidence
[44]
Rashid A. A 65-year-old man with anemia: diagnosis with peripheral blood smear. Blood research. 2015 Sep:50(3):129. doi: 10.5045/br.2015.50.3.129. Epub 2015 Sep 22
[PubMed PMID: 26457277]
[45]
DeLoughery TG. Iron Deficiency Anemia. The Medical clinics of North America. 2017 Mar:101(2):319-332. doi: 10.1016/j.mcna.2016.09.004. Epub 2016 Dec 8
[PubMed PMID: 28189173]
[46]
Kumar SB, Arnipalli SR, Mehta P, Carrau S, Ziouzenkova O. Iron Deficiency Anemia: Efficacy and Limitations of Nutritional and Comprehensive Mitigation Strategies. Nutrients. 2022 Jul 20:14(14):. doi: 10.3390/nu14142976. Epub 2022 Jul 20
[PubMed PMID: 35889932]
[47]
Kohne E. Hemoglobinopathies: clinical manifestations, diagnosis, and treatment. Deutsches Arzteblatt international. 2011 Aug:108(31-32):532-40. doi: 10.3238/arztebl.2011.0532. Epub 2011 Aug 8
[PubMed PMID: 21886666]
[48]
Aslinia F, Mazza JJ, Yale SH. Megaloblastic anemia and other causes of macrocytosis. Clinical medicine & research. 2006 Sep:4(3):236-41
[PubMed PMID: 16988104]
[51]
Zamani A, Kazerooni ES, Kasaee SS, Anbardar MH, Mohammadzadeh S, Shekarkhar G, Soleimani N. And the Oscar Goes to Peripheral Blood Film for the Detection of Lead Poisoning in a Complicated Toxic Patient: A Case Report with a Review of Laboratory Clues. Case reports in medicine. 2022:2022():9238544. doi: 10.1155/2022/9238544. Epub 2022 Feb 23
[PubMed PMID: 35251184]
Level 3 (low-level) evidence
[52]
Kano N, Fukui S, Kushiro S, Inui A, Saita M, Kura Y, Sawada U, Naito T. Basophilic stippling in red blood cells in the bone marrow: indication for lead poisoning diagnosis. The Journal of international medical research. 2022 Feb:50(2):3000605221078405. doi: 10.1177/03000605221078405. Epub
[PubMed PMID: 35184610]
[53]
Chwalba A, Maksym B, Dobrakowski M, Kasperczyk S, Pawlas N, Birkner E, Kasperczyk A. The effect of occupational chronic lead exposure on the complete blood count and the levels of selected hematopoietic cytokines. Toxicology and applied pharmacology. 2018 Sep 15:355():174-179. doi: 10.1016/j.taap.2018.05.034. Epub 2018 May 29
[PubMed PMID: 29857081]
[54]
Dobrakowski M, Boroń M, Czuba ZP, Birkner E, Chwalba A, Hudziec E, Kasperczyk S. Blood morphology and the levels of selected cytokines related to hematopoiesis in occupational short-term exposure to lead. Toxicology and applied pharmacology. 2016 Aug 15:305():111-117. doi: 10.1016/j.taap.2016.06.015. Epub 2016 Jun 11
[PubMed PMID: 27298078]
[55]
Porter M. Rapid Fire: Sickle Cell Disease. Emergency medicine clinics of North America. 2018 Aug:36(3):567-576. doi: 10.1016/j.emc.2018.04.002. Epub
[PubMed PMID: 30037443]