Continuing Education Activity
Bullous systemic lupus erythematosus (BSLE) is a rare manifestation of systemic lupus erythematosus. It has clinical and histological features that may lead to misdiagnosis and delayed treatment of this easily treatable condition. This activity reviews the evaluation and management of bullous systemic lupus erythematosus and highlights the role of the interprofessional team in improving care for patients with this condition.
Objectives:
- Outline the typical presentation of a patient with bullous systemic lupus erythematosus.
- Describe the histopathology and immunopathology of bullous systemic lupus erythematosus.
- Summarize management options available for bullous systemic lupus erythematosus.
- Review the importance of improving care coordination amongst the interprofessional team members to improve outcomes for patients affected by bullous systemic lupus erythematosus.
Introduction
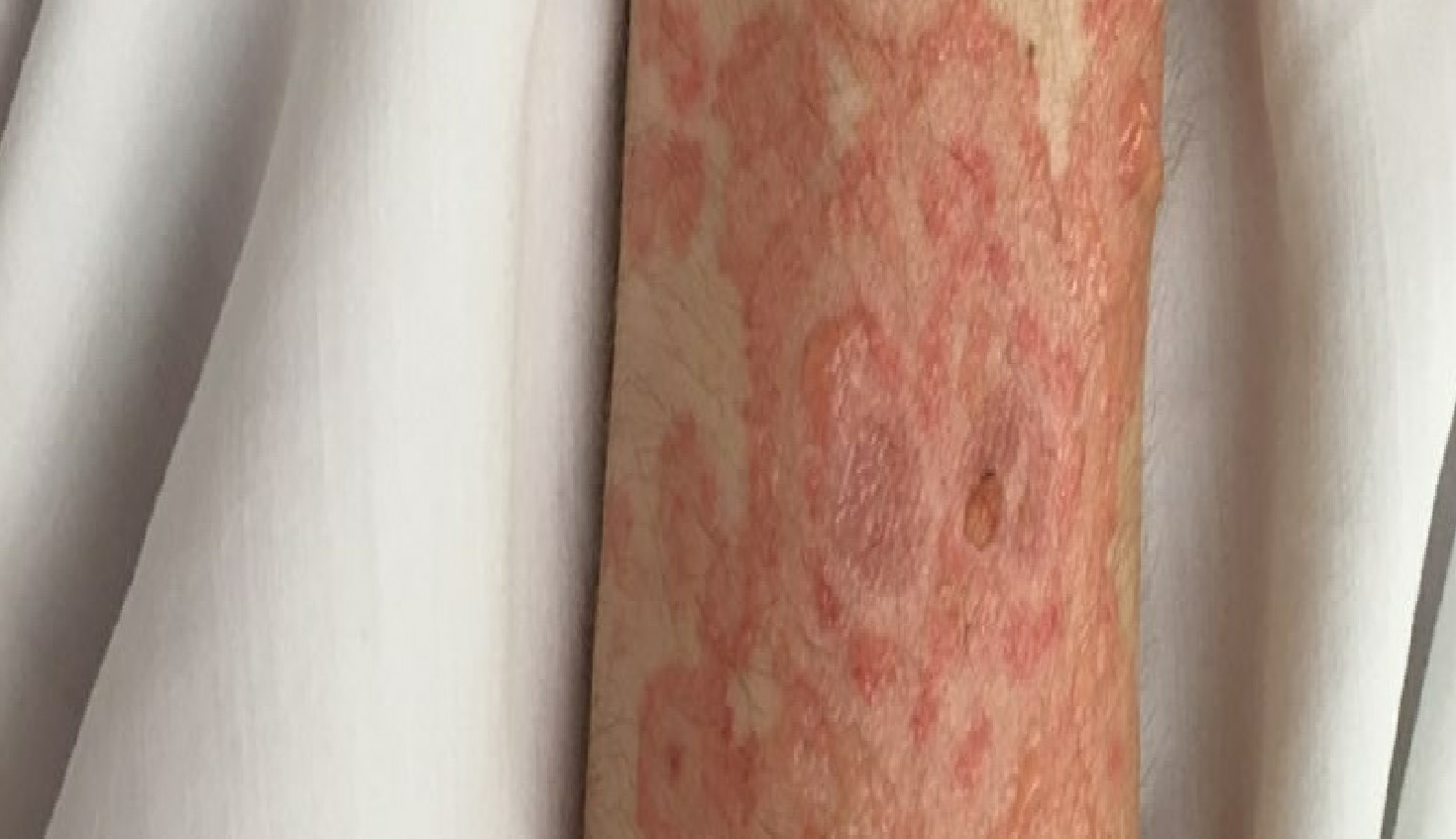
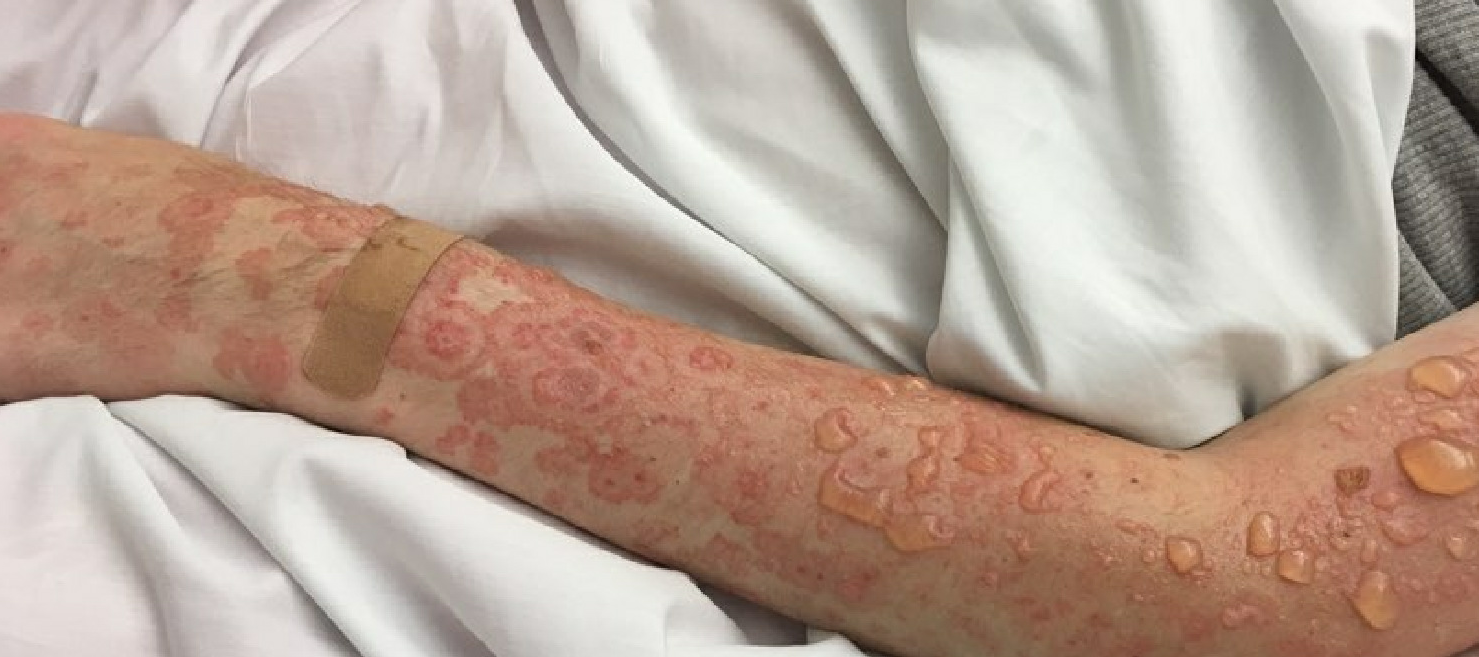
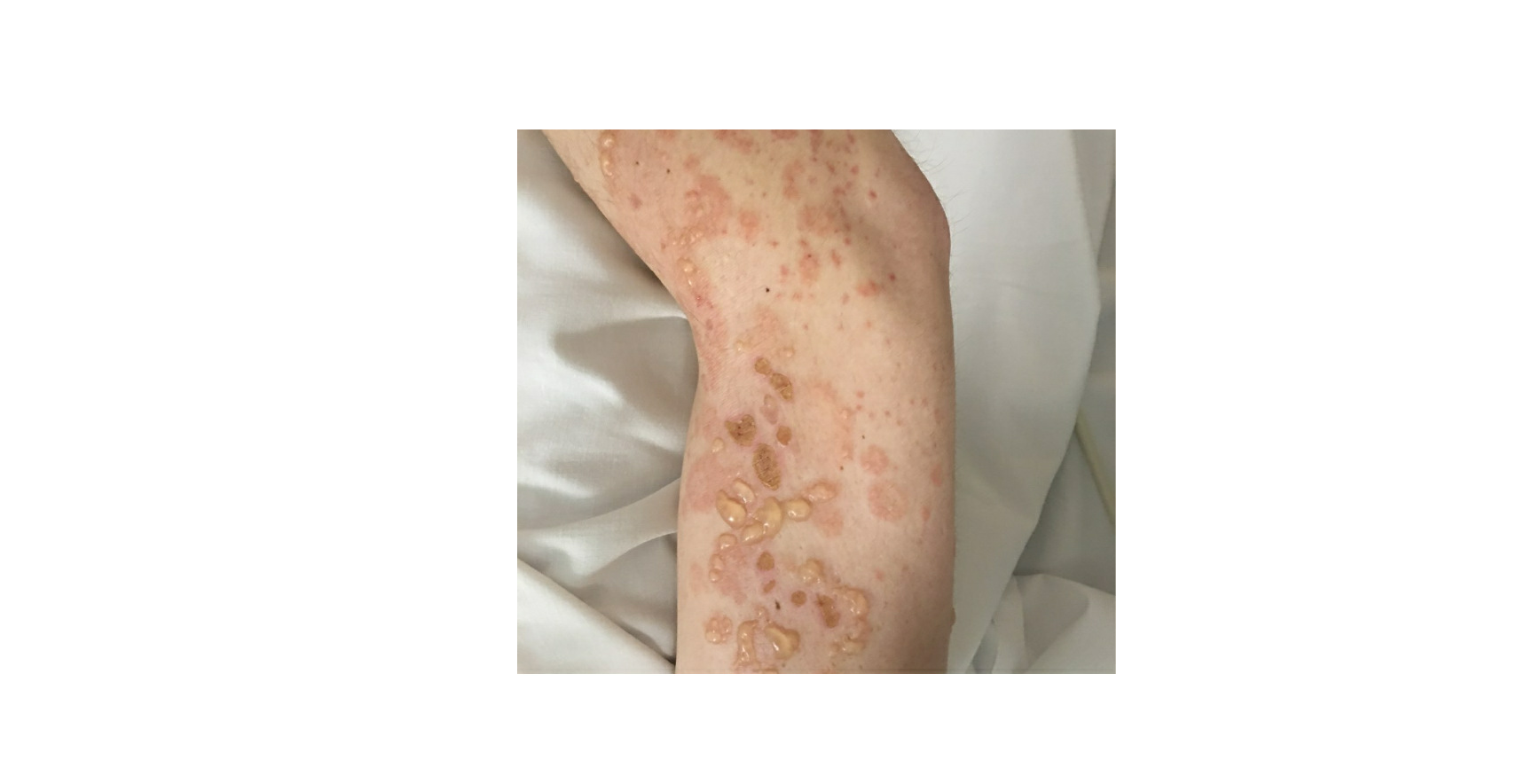
Lupus erythematosus is an autoimmune disease that has an array of clinical manifestations ranging from cutaneous to multi-organ systemic involvement. Bullous systemic lupus erythematosus (BSLE) is a rare blistering eruption seen in patients with systemic lupus erythematosus (SLE) described mainly in case reports, series, and a few multicenter retrospective studies. These cutaneous lesions appear as tense, vesiculobullous eruptions with a predilection for the extremities, trunk, face, and neck, usually healing without scar or milia.[1][2]
Etiology
The etiology of bullous systemic lupus erythematosus is unknown. It is an uncommon complication of SLE with autoantibodies to the dermo-epidermal junction of type VII collagen. Most patients with BSLE have an existing diagnosis of SLE, although there are several reports of BSLE as an initial presentation of SLE.[2][3]
Epidemiology
Bullous systemic lupus erythematosus, in line with the epidemiology of SLE, affects more females than males, usually of African descent in the second to the fourth decade of life. Nevertheless, it can be seen in all races, ages, or males. The reported incidence is limited but estimated at ~2% of all subepidermal autoimmune bullous cutaneous conditions.[4][5]
Pathophysiology
Autoantibodies to type VII collagen are involved in the pathophysiology of bullous systemic lupus erythematosus. This results in a weakened basement membrane-dermal adhesion, which appears as subepidermal blistering. Type VII collagen plays a vital role as an anchoring fibril attaching the epidermis to the dermis. The circulating autoantibodies are directed against the non-collagenous domain type 1 and 2 (NC1 and NC2) of type VII collagen seen in the basement membrane zone.[2][6] Other autoantibodies have been identified, such as BPAg1, laminin 5, laminin 6, and BPAg2.[7]
Histopathology
The histopathology of bullous systemic lupus erythematosus demonstrates subepidermal blistering with a dense neutrophilic infiltration in the upper dermis concentrated on the papillary tip with the association of nuclear dust and fibrin. These findings have a similarity to the histology of dermatitis herpetiformis. However, a good distinguishing feature is the presence of large deposits of mucin in the reticular dermis.[8][9]
Direct immunofluorescence (DIF) staining of perilesional and uninvolved skin shows the deposition of immune-reactants along the dermato-epidermal junction (DEJ). IgG constitutes the majority of immunoglobulins, but other classes such as IgA and IgM are seen. Complements are frequently seen in the involved skin. Granular, linear, or a mixed pattern of immune reactant deposition are seen in the basement membrane zone (BMZ). Indirect immunofluorescence staining performed with basement membrane zone-split skin may also be positive for antibodies to type VII collagen on the dermal side.[9] IgG subtyping can help differentiate the two.[10]
History and Physical
Bullous systemic lupus erythematosus is often an acute onset presentation with tense vesicles and bullae over erythematous or normal skin. These blisters appear more in sun-exposed areas but can be seen in non-sun-exposed skin and mucosa with a predilection for the trunk, face, neck, vermillion border, upper extremities-extensor surfaces, supraclavicular region, and oral mucosa.[4][11]
A rare pattern, erythema gyratum repens described as a centrifugally migrating erythematous plaque, has been described.[12][13]
BSLE may be associated with urticarial lesions, and pruritus is not always present. Healing with milia and/or scarring occurs infrequently. However, hypo/hyperpigmentation of the affected skin is usually seen.[8]
Several reports have shown an association with BSLE and active lupus nephritis. A multicenter retrospective study and literature review with a total of 128 cases described lupus nephritis in 50% of the cases, mainly class III or IV. 7% of cases had neuropsychiatric SLE and 45% with hematologic involvement at the time of BSLE.[8]
Evaluation
A careful review of the clinical presentation, serologies, histology, and immunofluorescence is required in the process of diagnosis.
In 1983 Camisa and Sharma proposed the following diagnostic criteria for bullous systemic lupus erythematosus, which was later revised in 1986:[14][15]
- A diagnosis of SLE based on the American College of Rheumatology (ACR) criteria
- Vesicles and bullae arising upon but not limited to sun-exposed skin
- Histopathology compatible with dermatitis herpetiformis (DH)
- Negative or positive indirect immunofluorescence (IIF) for circulating BMZ autoantibodies
- Direct immunofluorescence (DIF) revealing IgG and/or IgM and often IgA at the BMZ
In 1995, Yell et al. l described BSLE as "an acquired subepidermal blistering disease in a patient with SLE with immune reactants at the BMZ on either DIF or IIF."[11]
Detection of an autoantibody directed against collagen VII was shown to play a role in BSLE. The criteria by Camisa and Sharma were made before the availability of ELISA or immunoblot technology. The use of ELISA for detecting antibodies against NC1 and NC2 of type VII collagen is utilized.[16]
However, it is important to note that a diagnosis of SLE is usually present; however, this may be the first presentation of SLE in a few cases. A thorough exclusion of other blistering diseases should also be completed.
Treatment / Management
Dapsone is considered the first line of treatment with a good response.[17]
The use of dapsone, however, may have a few side effects, including but not limited to anemia, hepatitis, or hypersensitivity syndrome. In the event of intolerance, systemic manifestation, or extensive skin involvement, the use of steroids may be considered.
Several steroid-sparing agents have been used in the literature with variable efficacy. These immunosuppressive medications include cyclophosphamide, azathioprine, mycophenolate mofetil, methotrexate, with modest results. Rituximab has shown some promise with refractory cases.
Less infrequently, anakinra and intravenous immunoglobulins have been reported.[8]
Differential Diagnosis
Bullous systemic lupus erythematosus may be confused with several other blistering diseases, especially two autoimmune blistering conditions: dermatitis herpetiformis (DH) or epidermolysis bullosa acquisita (EBA).
Epidermolysis bullosa acquisita (EBA) exhibits similar antibodies against type VII collagen. It is more likely to heal with scarring and milia formation compared to BSLE. A prominent distinguishing feature includes the absence of SLE and a lack of histologic finding of large deposits of mucin in the dermis. The poor or slow response to dapsone in EBA compared to BSLE may help in separating the two entities.
Dermatitis herpetiformis (DH) has a skin distribution different from BSLE. The pruritic lesions have a predilection to the extensor surfaces, elbows, knees, and buttocks. DH is histologically identical to BSLE except for the presence of IgA deposition within the dermal papilla.[18]
Other differential diagnoses include:
- Linear IgA bullous dermatosis: immunofluorescence shows IgA deposition on the epidermal side of split skin
- Systemic lupus erythematosus with blisters
- Bullous pemphigoid: With the aid of immunofluorescence, antibody deposition is found on the epidermal side of salt-split skin
Prognosis
In a 12-year retrospective review, the majority of the patients healed without scarring or milia, although post-inflammatory hypo- or hyper-pigmentation may be seen. It was also pointed out that BSLE rarely recurred.[19]
Complications
Complications include:
- Infection of exposed and affected skin
- Sloughing esophagitis
- Complications from concomitant multiorgan SLE flare
- Medication side effects
Deterrence and Patient Education
Patient education is very vital in the management of bullous systemic lupus erythematosus. For a few, this may present as the first manifestation of SLE, which is a chronic, autoimmune condition with several multiorgan involvements. Educate these patients on the early signs and symptoms of organ involvement, photoprotection with sunscreen, and the need for medication adherence.
BSLE poses significant stress related to discomfort, aesthetics, and concern of skin complications. Support groups and behavioral therapy may be needed in such cases.
Enhancing Healthcare Team Outcomes
An interprofessional approach in evaluation, treatment, and care of patients with bullous systemic lupus erythematosus cannot be over-emphasized.
Primary care physicians, rheumatologists, and dermatologists play a key role in early diagnosis. The histopathology mimics of BSLE can be a source of a wrong diagnosis and delay of treatment, thus the need for an experienced pathologist.
Skincare by trained and dedicated nurses of the skin lesions, especially when ruptured leaving behind crusts and erosions, is also important.
Clinicians, nurses, and pharmacists also need to educate their patients on the disease condition, medications, and their potential side effects.
In cases with concomitant lupus nephritis, neuropsychiatric lupus, and hematologic complications, other subspecialties such as nephrology, neurology, and hematology may be needed.
Most importantly, the patient and family need to be involved in the treatment strategies, especially on decisions to escalate treatment to immunosuppressants. [Level 5]