Continuing Education Activity
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease with multisystem involvement and is associated with significant morbidity and mortality. Genetic, immunological, endocrine, and environmental factors influence the loss of immunological tolerance against self-antigens leading to the formation of pathogenic autoantibodies that cause tissue damage through multiple mechanisms. This activity reviews the evaluation and management of systemic lupus erythematosus and highlights the role of the interprofessional team in caring for patients with this condition.
Objectives:
- Identify when to consider systemic lupus erythematosus on the differential diagnosis.
- Describe the evaluation of systemic lupus erythematosus.
- Explain the treatment options for systemic lupus erythematosus.
- Review the importance of an interprofessional approach to enhance awareness, accurate diagnosis, and prompt treatment of systemic lupus erythematosus.
Introduction
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease with multisystemic involvement. The condition has several phenotypes, with varying clinical presentations from mild mucocutaneous manifestations to multiorgan and severe central nervous system involvement. Several immunopathogenic pathways play a role in the development of SLE. Hargraves described the lupus erythematosus (LE cell) in 1948. Several pathogenic autoantibodies have since been identified. Despite recent advances in technology and understanding of the pathological basis and risk factors for SLE, the exact pathogenesis is still not well known. Diagnosis of SLE can be challenging, and while several classification criteria have been posed, their utility in the clinical setting is still a matter of debate. Management of SLE is dictated by organ system involvement. Despite several agents shown to be efficacious in treating SLE, the disease still poses significant morbidity and mortality risk in patients.[1]
Etiology
SLE is a multisystemic disease with an unknown etiology. However, several genetic, immunological, endocrine, and environmental factors play a role in the etiopathogenesis of SLE.
Familial segregation and high concordance rates in identical twins suggest a strong genetic contribution in SLE, although there is no obvious inheritance pattern. Concordant rates for identical twins have been reported as high as 50%.[1] Over 100 gene loci with polymorphisms (or, rarely, copy numbers or mutations) have been identified to be associated with polygenic SLE (majority of cases), and more than 30 genes causing monogenic forms of SLE or SLE-like phenotype have been identified.[2] These genes are associated with activation of the immune system in response to foreign antigens, self-antigen generation, and activation of innate and adaptive immune systems. Some gene mutations that are rare but are considered very high risk for the development of SLE include deficiencies of early complement components C1q, C1r, C1s (>90% risk), C4 (50%), C2 (20%), and TREX1. Some of the other genes associated include HLA-DRB1, HLA-DR2, HLA-DR3, HLA-DRX, TNFAIP3, STAT-4, STAT-1, TLR-7, IRAK1/MECP2, IRF5-TNPO3, ITGAM, etc. The most common genetic predisposition is located at the major histocompatibility (MHC) locus. The MHC contains genes for antigen-presenting molecules (class I human leukocyte antigens [HLA-A, -B, and -C] and class II HLA molecules [HLA-DR, -DQ, and -DP]).[2]
In addition, women are at ten times more risk of developing SLE than men, and the risk of SLE is 14 times more in Klinefelter syndrome (47, XXY). This suggests an association with genes on the X-chromosome. However, despite several studies, the exact genes have not been identified.
Female sex and hormonal influence are significant risk factors for SLE. Estrogen stimulates CD8+ and CD4+ T cells, B cells, macrophages, thymocytes, the release of some specific cytokines (e.g., IL-1), and the expression of HLA and endothelial cell adhesion molecules (VCAM, ICAM). In addition, estrogens and prolactin promote autoimmunity, increase the B-cell activation factor production, and modulate lymphocyte and plasmacytoid dendritic cells (pDC) activation. The use of estrogen-containing contraceptives and postmenopausal hormone replacement therapy can cause flares in patients with SLE and have been associated with a higher incidence of SLE. In addition, elevated levels of prolactin are seen in patients with SLE. Androgens, on the other hand, are immunosuppressive.[3]
Several environmental triggers of SLE have been identified. Several drugs have been implicated in causing a lupus-like phenomenon by causing demethylation of DNA and alteration of self-antigens. While procainamide and hydralazine have the highest incidence of causing drug-induced lupus, more than 100 drugs have been associated with drug-induced lupus.[3] Further, several drugs such as the sulfa-drugs are well known to cause flares in patients with SLE. Ultraviolet rays and sun exposure lead to increased cell apoptosis and are well-known triggers for SLE. Several viral infections have been implicated, and the underlying mechanism is thought to be molecular mimicry. Antibodies against Epstein-Barr virus (EBV) are more prevalent in children and adults with SLE compared to the general population. Smoking is also thought to be a risk, with a dose-response.[4] Other potential risk factors include silica exposure, other viral infections, vitamin D deficiency, alfalfa sprouts, and foods containing canavanine.[4]
Epidemiology
Varying prevalence and incidence rates of SLE have been reported, with differences mainly attributed to the population differences. The Georgia and Michigan lupus registries reported prevalence of 72.1 to 74.4 per 100,000 persons and incidence rates of 5.6 per 100,000 person-years in primarily Caucasian and African-American populations. African-Americans have the highest rates, which are higher among Asian and Hispanic populations than Caucasians. The disease tends to have an earlier age of onset and is more severe in African-Americans.
SLE predominantly affects women of childbearing age, with a female to male ratio of 9 to 1. The risk, however, decreases after menopause in women, although still is twice as compared to men. Studies have indicated that although rare, lupus in men tends to be more severe. In addition, men tend to have more frequent skin manifestations, cytopenias, renal disease, serositis, neurologic involvement, thrombosis, cardiovascular disease, hypertension, and vasculitis than women.
Age also plays a vital role in SLE, and although the disease is more common in childbearing age in women, it has been well reported in the pediatric and elderly population. SLE is more severe in children SLE in children tends to be more severe than in adults, with a high incidence of malar rashes, nephritis, pericarditis, hematologic abnormalities, and hepatosplenomegaly. However, it tends to have a more insidious onset in older people and has more pulmonary involvement and serositis and less Raynaud's, malar rash, nephritis, and neuropsychiatric complications.[5]
Pathophysiology
The pathogenesis of SLE is complex, and the understanding of SLE pathogenesis is constantly evolving. A break in the tolerance in genetically susceptible individuals on exposure to environmental factors leads to the activation of autoimmunity. Cell damage caused by infectious and other environmental factors exposes the immune system to self-antigens leading to activation of T and B cells, which become self-sustained by a chronic self-aimed immune response. Cytokine release, complement activation, and autoantibody production lead to organ damage.
Both innate and adaptive immune systems play a role in the pathogenesis of SLE. The innate immune system activation is either Toll-like receptor (TLR) dependent or independent. The cell membrane-bound TLRs (TLR 2, 4, 6) are activated on exposure to the extracellular DNA and RNA from dying cells, which leads to downstream activation of the interferon regulatory family (IRF-3), NF-κB, and MAP-kinases, which serve as transcription factors for the production of proinflammatory mediators such as IFN-b. The endosomal TLRs (TLR 7, 9) are activated by single-stranded RNA and demethylated DNA, leading to interferon-alpha production and RNA binding autoantibodies such as antibodies against Ro La, Sm, and RNP. The TLR-independent pathway is activated by intracytoplasmic RNA sensors (RIG-1, MDA-5) and DNA sensors (IFI16, DAI) and leads to activation of IRF3 and NF-κB. Both self DNA/RNA and foreign DNA/RNA, such as from viruses, can lead to this activation. NETosis has recently gained attention in the pathogenesis of SLE. On activation by various factors such as cytokines, activated platelets, and vascular endothelial cells, neutrophils systematically release their nuclear aggregates in the extracellular environment. These nuclear aggregates can then promote Interferon-alpha production by the dendritic cells, mediate thrombosis and vascular damage and serve as self-antigens for T-lymphocytes.
T-lymphocytes and B-lymphocytes play a significant role in the pathogenesis of SLE. Apoptotic and damaged cell-derived antigens are presented to T-cells by antigen-presenting cells. T-cells in SLE display a distorted gene expression leading to the production of several cytokines. These T-cells produce less IL-2, which leads to altered regulatory T-cell production. Increased IL-6, IL-10, IL-12, and IL-23 increase mononuclear cell production while increased IL-17 and IL-21 lead to increased T-cel production. Increased Interfern-γ leads to defective T-cell production. T-cells lead to the activation of autoreactive B-cells by CD40L and cytokine production, leading to autoantibody production, a hallmark of SLE. Toll-like receptors on interaction with DNA and RNA lead to activation of these B-cells, and the nucleic acid and protein-containing intranuclear complexes are the most prominent antigens leading to B-cel activation. These autoantibodies are pathogenic and cause organ damage by immune complex deposition, complement, and neutrophil activation, altering cell function leading to apoptosis and cytokine production.[4][6]
Further, the autoreactive B-cells in SLE, stimulated by self-antigens, are not readily eliminated due to a deficiency of the process involved in the functional neutralization of autoreactive B cells. The B-cells can also serve as antigen-presenting cells and activate T-cells by presenting internalized soluble antigens to T-cells. This creates a loop where both B and T cells activate each other, leading to more autoimmunity.[7]
Histopathology
Tissue pathology in SLE can demonstrate a variety of aberrant immunologic mechanisms, including immune complex formation, autoantibody formation, and immunologically mediated tissue injury.
LE body or hematoxylin body is a hallmark of SLE pathology. It is a homogeneous globular mass of nuclear material that stains bluish-purple with hematoxylin. It can be observed in the lungs, kidneys, spleen, heart, lymph nodes, and serous and synovial membranes. They contain immunoglobulins and DNA, and phagocytes' engulfment of the LE body leads to the formation of the classic LE-cell.
Pathology from skin lesions in SLE demonstrates immune complex formation leading to tissue damage, vascular and perivascular inflammation, and chronic mononuclear cell infiltration. Acute lesions demonstrate fibrinoid necrosis at the dermo-epidermal junction and the dermis, along with liquefactive degeneration of the epidermis and perivascular inflammatory cell infiltration with a T-cell predominance. Chronic lesions can also demonstrate hyperkeratosis and follicular plugging. In addition, edema and RBC extravasation can be seen in all SLE lesions. Immunofluorescence demonstrates deposition of IgG, IgA, and IgM immunoglobulins and complement components along the dermal-epidermal junction.
Vasculitis is common in SLE, and vascular lesions may demonstrate various pathologies. Immune complex deposition with an inflammatory response is the most common lesion, although it may be seen without a significant inflammatory response. Small and large vessel necrotizing vasculitis with fibrinoid necrosis is less common but can be seen and differentiated from other vasculitides by immune complex deposition in the vessel wall. Thrombotic microangiopathy can present in patients with SLE and antiphospholipid antibody syndrome.[8]
In SLE, central nervous system pathology reveals small intracranial vessel involvement with thrombotic lesions with or without perivascular inflammation and endothelial proliferation. Necrotizing vasculitis is present rarely. Thromboembolism from Libman-Sacks endocarditis has been seen as well.
Cardiac pathology may include valvular involvement leading to Libman-Sacks endocarditis which is sterile verrucous endocarditis. It tends to involve the mitral valve, most commonly with vegetations seen on the forward flow side of the valve. Pathology reveals platelet thrombi, necrotic cell debris, proteinaceous deposits, and mononuclear cells. Pericarditis with fibrinous exudate is common, and pathology reveals mononuclear cells' fibrinoid necrosis and perivascular infiltration. Myocarditis can be seen as well. SLE poses a very high risk for atherosclerotic coronary artery disease, and vasculitis, immune complex deposition in addition to corticosteroid use, and hypertension are thought to be contributory.[9]
Lymphadenopathy is common in SLE, and pathology may reveal follicular hyperplasia with giant cells, plasma cells infiltration of the interfollicular zones, and necrosis of the paracortical T-cell zones. LE bodies may be rarely seen. The necrotic vessel wall shows immunoglobulin and Complement C3 deposition. Splenomegaly is also common in SLE, with pathology showing the classic onionskin lesion with has multiple concentric rings of perivascular collagen. Follicular hyperplasia and periarterial fibrosis are common.
Lupus pneumonitis can be seen in up to 10% of lupus patients. Interstitial pneumonitis, alveolitis, alveolar wall injury, and edema and hemorrhage are commonly seen in these patients. Immunoglobulin and complement deposition is seen in the vessel wall. Chronic interstitial lung disease can occur in up to 50% of these patients and is characterized by interstitial lymphoid aggregates and fibrosis, septal thickening, and type-2 pneumocyte hyperplasia. Medial hypertrophy and intimal fibrosis involving the pulmonary artery branches lead to pulmonary hypertension in SLE. Again, immunoglobulin and complement deposition can be seen in the vessel wall.
Lupus nephritis can involve the glomeruli, interstitium, tubules, and vessels with immune complex deposition in all four compartments. The World Health Organization classification criteria for lupus nephritis describes six classes of lupus nephritis, all with distinct pathological features and significant differences in clinical outcomes. This has led to a different treatment approach for each class and knowing the class of lupus nephritis before initiating treatment is vital.
- Class I: Minimal mesangial lupus nephritis
- Class II: Mesangial proliferative lupus nephritis
- Class III: Focal lupus nephritis
- Class IV: Diffuse segmental or Diffuse global lupus nephritis
- Class V: Membranous lupus nephritis
- Class VI: Advanced sclerosing lupus nephritis[10]
History and Physical
SLE is a multisystem disease with several phenotypes. Clinical features may vary from a very mild disease with only mucocutaneous involvement to severe life-threatening disease with multiorgan involvement. All organ systems can be involved in SLE. An autoantibody profile can sometimes help predict the disease course and clinical features. Several studies have indicated the development of serological abnormalities several years before the onset of clinical lupus. This is termed pre-clinical lupus, where a patient may have serological abnormalities consistent with SLE and may have some clinical features but still does not meet the criteria for SLE. There is evidence that a significant percentage of these patients with pre-clinical lupus, including those with incomplete lupus or undifferentiated connective tissue disease, may transition to clinical lupus and fulfill the SLE criteria later in life.
Constitutional Symptoms
Constitutional symptoms are seen in more than 90% of patients with SLE and are often the initial presenting feature. Fatigue, malaise, fever, anorexia, and weight loss are common. While more than 40% of patients with SLE may have lupus flare as a cause of fever, infections must always be ruled out first, given the immunocompromised state of these patients. Further, SLE is an infrequent cause of fever of unknown origin.[11]
Mucocutaneous Manifestations
More than 80% of patients with SLE suffer from mucocutaneous involvement, one of the most well-known and identified clinical features. SLE skin lesions may be lupus specific, while several nonspecific lesions are also seen. Lupus-specific lesions include (1). Acute cutaneous lupus erythematosus (ACLE) includes localized, malar, and generalized, (2) Subacute cutaneous lupus erythematosus (SCLE) includes annular and papulosquamous, and (3) Chronic cutaneous lupus erythematosus (CCLE) includes classic discoid lupus erythematosus (DLE), hypertrophic/verrucous, lupus panniculitis/profundus, lupus tumidus, chilblains lupus, mucosal discoid lupus, and lichenoid discoid lupus.
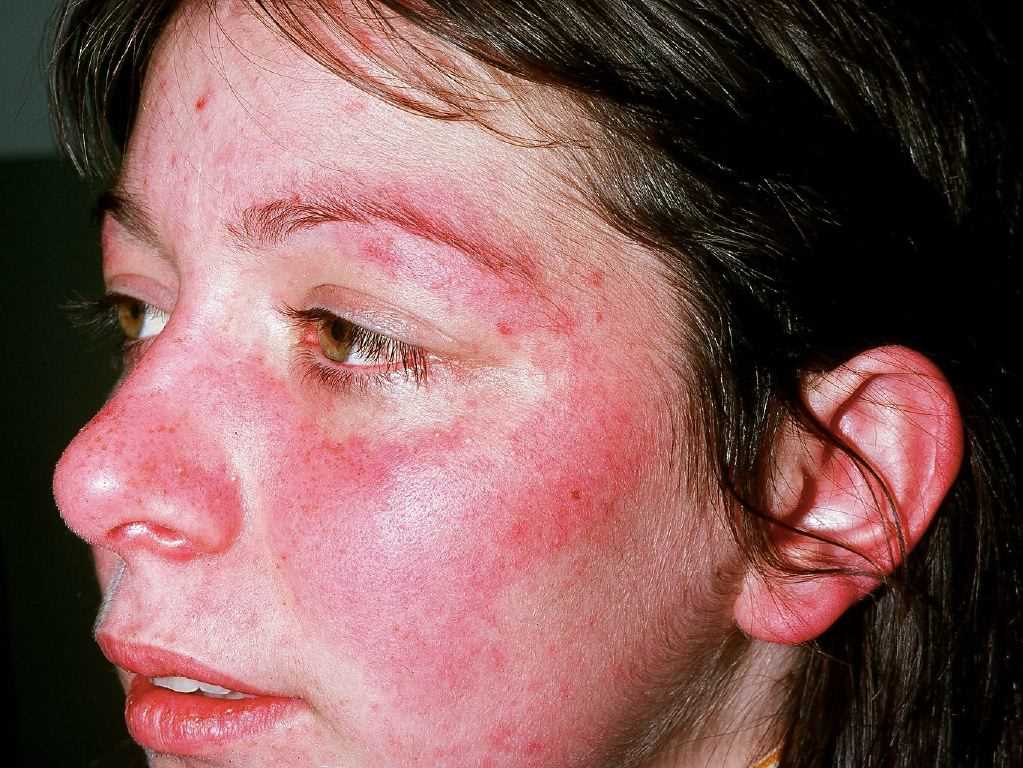
Acute cutaneous lupus erythematosus (ACLE) may be localized or generalized. The hallmark ACLE lesion is the malar rash or the butterfly rash, an erythematous raised pruritic rash involving the cheeks and nasal bridge. The rash may be macular or papular and spares the nasolabial folds (photoprotected). It usually has an acute onset but may last several weeks and cause induration and scaling. The malar rash may also fluctuate with lupus disease activity. Other rashes in this location that must be differentiated from ACLE malar rash include rosacea, erysipelas, seborrheic dermatitis, and perioral dermatitis. Generalized ACLE leads to widespread maculopapular or macular rash in a photosensitive pattern. ACLE lesions usually heal without scarring.
Subacute cutaneous lupus erythematosus (SCLE) rash is a photosensitive, widespread, nonscarring, nonindurated rash. SCLE may be either papulosquamous resembling psoriasis or an annular/polycystic lesion with central clearing and peripheral scaling. SCLE lesions may last several months but usually, heal without scarring. SCLE rash is seen in patients with a positive Anti-Ro (SSA) antibody in up to 90% of the cases. SCLE can also be caused by some drugs such as hydrochlorothiazide.[3] It has also been reported in patients with Sjogren syndrome and rheumatoid arthritis.[4]
Discoid lupus erythematosus (DLE) is the most common form of chronic cutaneous lupus erythematosus (CCLE). DLE may occur with or without SLE and can be localized (only head and neck) or generalized (above and below the neck). The lesions are disk-shaped erythematous papules or plaques with adherent scaling and central clearing. DLE heals with scarring and can be associated with permanent alopecia when present on the scalp. Mucosal DLE lesions can be seen in the oral cavity, and these tend to be painful erythematous round lesions with white radiating hyperkeratotic striae. Hypertrophic DLE may mimic squamous cell carcinoma histologically. Lupus panniculitis can occur above the waist and is less likely associated with SLE. The lesions result in depressed areas, and when associated with DLE lesions overlying them, they are known as lupus profundus. Chilblains lupus presents with erythematous tender plaques on fingers and toes. Lupus tumidus lesions are erythematous, edematous smooth plaques without epidermal involvement.
Oral and nasal ulcers are common in SLE, and acutely, often are painless. They may present as gradual onset erythema, macule, petechiae, erosions, or ulcers involving any part of the oral cavity. The most common locations are the hard palate, the buccal mucosa, and the vermilion border. Photosensitivity is present in SLE in more than 90% of cases. It is characterized by abnormal skin reaction on exposure to ultraviolet A/B and visible light, a reaction that may last weeks to months. These patients also experience worsening of their systemic symptoms on sun exposure. Alopecia in SLE may be due to DLE (scarring), or the brittle, easily breaking lupus hair (non-scarring) in the temporal/parietal area.
Several other skin manifestations are seen in SLE that are not specific. These include cutaneous vasculitis (leukocytoclastic or urticarial), vasculopathy (livedo reticularis, superficial thrombophlebitis, Raynaud's phenomenon, erythromelalgia, periungual telangiectasia), sclerodactyly, rheumatoid nodules, calcinosis cutis, bullous lesions, urticaria, erythema multiforme, acanthosis nigricans, lichen planus, and leg ulcers.[12]
Musculoskeletal Manifestations
Approximately 80 to 90% of patients with SLE suffer from musculoskeletal involvement at some point during their disease course and may range from mild arthralgias to deforming arthritis. Lupus arthritis is typically a non-erosive, symmetrical inflammatory polyarthritis affecting predominantly the small joints of the hands, knees, and wrists, although any joint can be involved. Jaccoud arthropathy results from the joint capsule and ligament laxity, leading to non-erosive deformities of the hands, including ulnar deviation and subluxation of the metacarpophalangeal joints that may mimic rheumatoid arthritis. Usually, these deformities are reducible, although rarely, they may become fixed. Avascular necrosis (with or without steroid use) can occur in up to 10% of patients with SLE and is usually bilateral and involves the hip joints. Inflammatory myopathy with histopathological features similar to but less striking than polymyositis has been seen in less than 10% of SLE cases. Patients with SLE are at high risk for the development of fibromyalgia, with incidences as high as 20% reported. Rheumatoid nodules have been reported in patients with SLE.[13]
Hematologic and reticuloendothelial manifestations:
Anemia is present in more than 50 % of patients with SLE and most commonly is anemia of chronic disease. Other causes of anemia in SLE may include iron deficiency anemia, coomb's positive autoimmune hemolytic anemia, red blood cell aplasia, and microangiopathic hemolytic anemia, which may be associated with antiphospholipid antibody syndrome. Leukopenia secondary to neutropenia or lymphopenia is also very frequent and severe. Thrombocytopenia can be mild or severe and may be associated with antiphospholipid antibody syndrome and autoantibodies against platelets, glycoprotein IIb/IIIa, or thrombopoietin receptor. Pancytopenia is not infrequent and may occasionally be associated with myelofibrosis. Soft non-tender lymphadenopathy is common in SLE, although rare cases of histiocytic necrotizing lymphadenitis have been reported (Kikuchi-Fujimoto disease). Splenomegaly is common in SLE, while splenic atrophy and asplenism have been reported.
Neuropsychiatric Manifestations
Both central (CNS) and peripheral (PNS) nervous systems may be involved in SLE in addition to several psychiatric manifestations, although the diagnosis can be difficult. The most common CNS manifestation is intractable headaches, reported in more than 50% of cases. Focal or generalized seizures may be seen, and are associated with disease activity, although they carry a favorable prognosis. Other CNS manifestations include aseptic meningitis, demyelinating syndrome including optic neuritis and myelitis, movement disorders such as chorea and cognitive dysfunction. Patients with SLE are also at high risk for ischemic strokes. Cranial and peripheral (sensorimotor, axonal) neuropathies, mononeuritis multiplex, autonomic neuropathies, and syndromes mimicking Guillain-Barré syndrome and Myasthenia gravis are the peripheral nervous system manifestations. Psychiatric manifestations are difficult to diagnose and manage and may range from depression and anxiety to frank psychosis.
Renal Manifestations
Lupus nephritis is a well-known and common complication of SLE. The involvement may range from mild subnephrotic proteinuria to diffuse progressive glomerulonephritis leading to chronic kidney damage. Lupus nephritis usually occurs early in the course of SLE. New-onset hypertension, hematuria, proteinuria, lower extremity edema, and elevation in creatinine shall raise suspicion for lupus nephritis. A biopsy is crucial in staging lupus nephritis and ruling out other causes. The six classes of lupus nephritis are mentioned in the histopathology section of this article. The biopsy findings dictate the treatment of lupus nephritis. The prognosis varies for each class, with an excellent prognosis for classes I and II and poor outcomes with classes III and IV. Class V usually carries a favorable prognosis except for complications of nephritis syndrome such as thromboembolism which are common in this class. Other renal manifestations may include thrombotic microangiopathy, interstitial nephritis, lupus vasculopathy, vasculitis, and arteriolosclerosis.[14]
Pulmonary Manifestations
Pleuritis is the most common pulmonary manifestation and may not always be associated with pleural effusion. Other pulmonary manifestations include exudative pleural effusions, acute lupus pneumonitis with bilateral pulmonary infiltrates, an interstitial lung disease which may be nonspecific interstitial pneumonia (NSIP) or usual interstitial pneumonia (UIP), diffuse alveolar hemorrhage associated with capillaritis, pulmonary arterial hypertension, pulmonary embolism (with or without antiphospholipid antibody syndrome) and shrinking lung syndrome.
Cardiovascular Manifestations
SLE may involve any layer of the heart, including the pericardium, myocardium, endocardium, and even the coronary arteries. Pericarditis associated with exudative pericardial effusions is the most common cardiac manifestation. Cardiac tamponade is rare. Myocarditis is rare and is associated with anti-Ro (SSA) antibodies. Hydroxychloroquine-associated cardiomyopathy shall be ruled out, and this may occasionally require an endomyocardial biopsy. Valvular abnormalities, including Libman-Sacks endocarditis involving the mitral valve, are common and may be associated with antiphospholipid antibody syndrome. Patients with SLE are especially at high risk for coronary artery disease, either due to coronary vasculitis or more frequently due to generalized atherosclerosis.[4]
Gastrointestinal Manifestations
Any part of the gastrointestinal tract may be involved in SLE. These manifestations include esophageal dysmotility (especially the upper one-third of the esophagus), mesenteric vasculitis, lupus enteritis, peritonitis and ascites, protein-losing enteropathy, pancreatitis, and lupoid hepatitis. Further, patients with SLE and antiphospholipid antibody syndrome can develop Budd-Chiari syndrome, mesenteric vessel thrombosis, and hepatic veno-occlusive disease.
Pregnancy Complications
SLE patients with positive antiphospholipid antibodies are at a high risk of spontaneous abortions and fetal loss, pre-eclampsia, and maternal thrombosis. Anti-Ro (SSA) and Anti-La (SSB) antibodies can cross the placenta leading to fetal heart block and neonatal lupus presenting with a photosensitive rash, cytopenias, and transaminitis. The risk is 2% with the first pregnancy but increases to 20% if there is a history of neonatal lupus in a past pregnancy. SLE usually flares in pregnancy, especially if the disease was uncontrolled in the six months preceding pregnancy. Lupus nephritis can be challenging to differentiate from pre-eclampsia, although several clinical and laboratory features (low complements, positive Anti-Ds-DNA antibody, normal serum uric acid level, and active urinary sediment) may help. Patients with more severe SLE manifestations such as pulmonary hypertension, severe cardiovascular disease, or cerebrovascular accident are especially at a very high risk of mortality during pregnancy.[15]
Other Manifestations
Eye involvement is common, and keratoconjunctivitis sicca is frequently seen in SLE in the presence or absence of secondary Sjogren syndrome. Other ocular manifestations are retinal vasculitis, optic neuritis, uveitis, scleritis, peripheral ulcerative keratitis, and episcleritis. Patients with SLE are also more susceptible to drug-induced ocular damage, including steroid-induced glaucoma or cataract and hydroxychloroquine-induced maculopathy. Ear involvement may lead to sudden sensorineural hearing loss. Adrenal infarction secondary to adrenal vessel thrombosis may be seen in patients with SLE and antiphospholipid antibody syndrome.
Classification Criteria
American College of Rheumatology (ACR) first developed the SLE classification criteria in 1971 and revised them in 1982 and 1997. The 1997 ACR criteria were further revised by the Systemic Lupus International Collaborating Clinics (SLICC) group in 2012.
The 1997 ACR criteria required 4 out of 11 criteria for the classification of SLE. The 11 criteria included were malar rash, discoid rash, photosensitivity, alopecia, Raynaud phenomenon, oral/nasal ulcers, arthritis (non-erosive arthritis involving 2 or more peripheral joints), serositis (pleurisy or pericarditis), renal disease (proteinuria greater than 500 mg daily or cellular RBC, granular, tubular, or mixed casts), hematologic disease (hemolytic anemia with reticulocytosis, or leukopenia less than 4000/mm3 on 2 or more occasions or lymphopenia less than 1500/mm3 on 2 or more occasions, or thrombocytopenia less than 100,000/mm3 in the absence of medications known to decrease platelets), neurologic disease (seizures or psychosis in the absence of an alternative explanation), immunologic criteria (Antiphospholipid antibodies present based on either an abnormal serum level of IgM or IgG anticardiolipin antibodies or a tested positive result for lupus anticoagulant or anti-DNA antibody or anti-Sm antibody or false-positive syphilis test with VDRL or RPR) and antinuclear antibody positivity in the absence of drugs known to cause drug-induced lupus.
The Systemic Lupus International Collaborating Clinics (SLICC) criteria made notable changes to the 1997 ACR to improve clinical relevance. It requires at least one of the four criteria to be clinical and at least one of the four criteria to be immunologic. In addition, neurologic and immunologic criteria were expanded to include new information about SLE immunology. Further, patients with biopsy-proven nephritis and positive ANA or anti-double-stranded DNA could be directly classified as SLE even if they lacked any other criteria. Compared to the 1997 ACR criteria, the SLICC criteria have improved sensitivity and are considered more valid and clinically relevant.
In September 2019, the European League Against Rheumatism (EULAR) and the American College of Rheumatology (ACR) published new criteria for the classification of SLE. The EULAR/ACR criteria have a specificity of 93.4% and sensitivity of 96.1%, as opposed to 93.4% specificity and 82.8% sensitivity of the 1997 ACR criteria, and 83.7% specificity and 96.7% sensitivity of the 2012 Systemic Lupus International Collaborating Clinics (SLICC) classification criteria. Each criterion is assigned points, from 2 to 10. Patients with 10 or more points and at least one clinical criterion are classified as having SLE.[16]
Table 1. EULAR/ACR Clinical Domains and Criteria for SLE
|
Domain
|
Criteria
|
Points
|
|
Constitutional
|
Fever
|
2
|
|
Hematologic
|
Leukopenia
Thrombocytopenia
Autoimmune hemolysis
|
3
4
4
|
|
Neuropsychiatric
|
Delirium
Psychosis
Seizure
|
2
3
5
|
|
Mucocutaneous
|
Non-scarring alopecia
Oral ulcers
Subacute cutaneous or discoid lupus
Acute cutaneous lupus
|
2
2
4
6
|
|
Serosal
|
Pleural or pericardial effusion
Acute pericarditis
|
5
6
|
|
Musculoskeletal
|
Joint involvement
|
6
|
|
Renal
|
Proteinuria > 0.5 g/24 h
Renal biopsy class II or V lupus nephritis
Renal biopsy class III or IV lupus nephritis
|
4
8
10
|
Table 2. EULAR/ACR Immunologic Domains and Criteria for SLE
|
Domain
|
Criteria
|
Points
|
|
Antiphospholipid antibodies
|
Anticardiolipin antibodies or
Anti-β2GP1 antibodies or
Lupus anticoagulant
|
2
|
|
Complement proteins
|
Low C3 or low C4
Low C3 and low C4
|
3
4
|
|
SLE-specific antibodies
|
Anti-dsDNA antibody or
Anti-Smith antibody
|
6
|
It must be noted that these criteria are developed for research studies for the classification of patients and may not always be valid in the clinical setting. Therefore, while they can assist a physician in suspecting a diagnosis of SLE, they alone shall not be considered enough in confirming or ruling out a diagnosis of SLE, which is still a clinical diagnosis made by an expert while considering the whole clinical presentation along with serological and histopathological testing, and imaging.
Evaluation
The diagnosis of SLE can be challenging, and no single clinical feature or lab abnormality can confirm SLE diagnosis. Instead, SLE is diagnosed based on the constellation of signs, symptoms, and appropriate laboratory workup. Imaging and histopathology may play a crucial role as well.
Several autoantibodies have been described in SLE, with varying sensitivity and specificity. While some autoantibodies may be associated with a certain clinical subset of SLE, others may serve as a marker of disease activity.
Antinuclear antibodies (ANA) are the hallmark of the disease and shall be the initial test performed. Immunofluorescence assay is considered the gold standard test for ANA. Although other detection methods such as enzyme-linked immunosorbent assay (ELISA) and multiplex assays are widely available, they lack sensitivity. A positive ANA is seen in more than 97% of cases of SLE. However, it can also be seen in several other disorders and a significant proportion of the healthy population, and have a specificity of only 20%. Hence, a positive ANA does not confirm SLE diagnosis, but a negative ANA makes it significantly less likely. ANA negative SLE has been rarely described, although it is primarily due to methodical error. Those cases have either a positive ANA on immunofluorescence or a positive Anti-Ro (SSA) antibody.
Several patterns of ANAs have been reported, including speckled, homogenous, centromere, cytoplasmic, nucleolar, and dense fine speckled patterns. With the availability of more specific ANAs targeting specific antigens, the staining patterns of ANAs are not considered significant enough by themselves. ANAs with a dense, fine speckled pattern (anti-DFS70) are considered least pathological, and patients with ANAs with this pattern rarely develop systemic autoimmune diseases. The speckled pattern is seen when ANAs are directed against the antigens such as SSA, SSB, Smith, ribonucleoprotein. The homogenous pattern is associated with ANAs targeted at histones, chromatin, and dsDNA, while the centromere pattern is associated with Anti-centromere antibodies seen in limited systemic sclerosis.[17]
Besides SLE, ANAs can be seen in several other conditions, as noted above. More than 20% of the healthy normal population, especially females or relatives of patients with autoimmune diseases, can have a positive ANA, although titers more than 1:320 are uncommon. Other rheumatological disorders associated with a positive ANA include drug-induced lupus, systemic sclerosis, polymyositis/dermatomyositis, mixed connective tissue disease, Sjogren syndrome, rheumatoid arthritis, juvenile idiopathic arthritis, Raynaud's disease, cutaneous lupus, and fibromyalgia. Several other autoimmune disorders are associated with a positive ANA, including autoimmune hepatitis, multiple sclerosis, Hashimoto thyroiditis, and idiopathic thrombocytopenic purpura. Several infections and malignancies have also been associated with a positive ANA.
A positive ANA shall be followed by testing for more specific autoantibodies to detect the antigen responsible for the positive ANA. It must be noted that frequently, a positive ANA will not be associated with any of the known more specific autoantibodies. There are several possible targets for ANAs, with any peptide synthesized inside the cell's nucleus serving as a potential antigen. However, only a few have been identified as having clinical relevance so far. A positive ANA with negative testing for more specific autoantibody testing is less likely to be associated with systemic autoimmune disease.
Antibodies to deoxyribonucleic acid (DNA) can be primarily divided into two groups: those reactive with denatured, single-stranded (ss)DNA and those identifying native, double-stranded (ds)DNA. Notably, anti-ssDNA antibodies are considered non-specific and may be seen either as a laboratory error or in the healthy population. Anti-double-stranded deoxyribonucleic acid (dsDNA) antibodies have more than 95% specificity for SLE but are found in only about 60% to 70% of SLE patients. Thus a negative anti dsDNA does not rule out the diagnosis of SLE. The Farr radioimmunoassay test is considered the gold standard for detecting anti-dsDNA antibodies, although it is not frequently used. ELISA tests are available, but they have a high risk of giving a false positive test. The immunofluorescence test by using the Crithidia luciliae method can confirm the presence of anti-Ds-DNA antibodies. Anti-dsDNA antibodies can also be seen in drug-induced lupus, primarily secondary to anti-TNF agents and interferon-alpha. Rarely low titers of anti-dsDNA antibodies have been reported in rheumatoid arthritis and Sjogren syndrome. In SLE, anti-dsDNA antibodies can correlate with disease activity and the development of lupus nephritis. However, this may not always be true as some patients have elevated anti-dsDNA antibodies in the setting of minimally active or inactive lupus.
Anti-Ro (SSA) and anti-La (SSB) antibodies target ribonucleoprotein particles. Anti-Ro and Anti-La antibodies are seen in up to 90% of cases of Sjogren syndrome but can be seen in SLE as well (anti-Ro in up to 50% and anti-La in up to 20% of the cases). In SLE, they may be associated with secondary Sjogren syndrome and keratoconjunctivitis sicca, subacute cutaneous lupus, photosensitivity, congenital heart block, and neonatal lupus.[18]
Anti-Smith antibodies are seen in less than 30% of SLE patients but have 99% specificity for SLE. They are observed more in African-American patients with SLE. Anti-Smith antibodies in SLE are usually always associated with Anti-U1-RNP antibodies, which are present in up to 30% of SLE patients. Anti-U1-RNP antibodies can also be seen in mixed connective tissue disease (MCTD), although in MCTD, a disorder that is closely related to SLE. Anti-ribosomal-P antibodies are very specific for SLE, although their prevalence in SLE is less than 5%, and they may correlate with neuropsychiatric manifestations of SLE. Anti-histone antibodies are not specific for drug-induced lupus and can be seen in 50% to 70% of cases of SLE. Anti-centromere and anti-topoisomerase-I (SCL70) antibodies are seen in systemic sclerosis and rarely in SLE (less than 5%). Anti-histidyl-tRNA-synthetase antibodies are seen in myositis. Patients with SLE may also have antiphospholipid antibodies (lupus anticoagulants, anti-cardiolipin, and anti-beta-2-glycoprotein I antibodies) and are associated with more thrombotic events and adverse pregnancy-related outcomes.
Complements C3 and C4 shall be checked in patients with SLE or suspicion of SLE, and low complement levels indicate complement consumption and may correlate with disease activity. Markers of inflammation such as erythrocyte sedimentation rate and C-reactive protein may be elevated. Complete blood counts, liver function tests, and renal function tests, including serum creatinine, urinalysis, and urine protein quantification (24-hour urine protein, or spot urine protein/creatinine ratio), shall be checked to assess organ involvement. Synovial fluid aspiration reveals an inflammatory fluid. Joint radiographs may demonstrate peri-articular osteopenia, deformities, or subluxation but rarely show erosions. Chest imaging with computed tomography (CT) scan, cardiac workup including echocardiography (trans-esophageal when suspecting Libman-Sacks endocarditis), CNS work up with magnetic resonance imaging (MRI), and/or lumbar puncture shall be pursued if specific organ involvement is suspected. Renal biopsy shall always be performed if suspicion of lupus nephritis. Skin biopsies can be considered, especially if atypical presentation.[19]
Treatment / Management
Treatment in SLE aims to prevent organ damage and achieve remission. The choice of treatment is dictated by the organ system/systems involved and the severity of involvement and ranges from minimal treatment (NSAIDs, antimalarials) to intensive treatment (cytotoxic drugs, corticosteroids).
Patient education, physical and lifestyle measures, and emotional support play a central role in managing SLE. Patients with SLE shall be well educated on the disease pathology, potential organ involvement, including brochures, and the importance of medication and monitoring compliance. Stress reduction techniques, good sleep hygiene, exercises, and emotional support shall be encouraged. Smoking can worsen SLE symptoms, and patients should be educated about the importance of smoking cessation. Dietary recommendations shall include avoiding alfalfa sprouts and echinacea and including a diet rich in vitamin D. Photoprotection is vital. All patients with SLE shall avoid direct sun exposure by timing their activities appropriately, light-weight loose-fitting dark clothing covering the maximum portion of the body, and using broad-spectrum (UV-A and UV-B) sunscreens with a sun protection factor (SPF) of 30 or more.
Cutaneous manifestations: Mild cutaneous manifestations can usually be treated with topical corticosteroids or topical calcineurin inhibitors such as tacrolimus. Hydroxychloroquine is the drug of choice for most cutaneous manifestations and is very efficacious. Quinacrine can be used if intolerance or adverse effects of hydroxychloroquine. Methotrexate can be used if no response to hydroxychloroquine. For severe or resistant disease, systemic corticosteroids, mycophenolate mofetil (dual-benefit with underlying lupus nephritis), and belimumab can be considered. Other alternatives include thalidomide, cyclophosphamide, IVIG, dapsone, azathioprine, and rituximab.[20]
Musculoskeletal manifestations: Hydroxychloroquine is the initial drug of choice for lupus arthritis. If no response, methotrexate or leflunomide can be considered. Belimumab and rituximab can be considered in refractory cases.[21]
Hematological manifestations: Drug-induced cytopenias shall be excluded. Mild cytopenias usually require no treatment. For moderate to severe cytopenias, corticosteroids are the mainstay of treatment, and azathioprine or cyclosporine-A can be used as a steroid-sparing agent. Severe refractory cytopenias may require intravenous pulse dose steroids, mycophenolate mofetil, rituximab, cyclophosphamide, plasmapheresis, recombinant G-CSF, or splenectomy.[22]
Cardiopulmonary manifestations: Serositis usually responds to NSAIDs or moderate to high dose oral corticosteroids. Hydroxychloroquine and methotrexate can be considered as steroid-sparing agents. Acute lupus pneumonitis requires high dose IV pulse corticosteroids, while plasmaphereses and/or cyclophosphamide may be needed if the diffuse alveolar hemorrhage is present. Interstitial lung disease can e managed by low to moderate dose corticosteroids with immunosuppressive agents such as azathioprine or mycophenolate mofetil. Pulmonary arterial hypertension requires vasodilator therapy, while thrombotic complications such as pulmonary embolism require anticoagulation. Therefore, high-dose corticosteroids are necessary to manage myocarditis and coronary arteritis.
CNS manifestations: Accurate diagnosis and ruling out other potential causes is critical before initiating treatment for neuropsychiatric manifestations of SLE. High-dose corticosteroids with immunosuppressive agents such as cyclophosphamide, azathioprine, or rituximab are used for inflammation-related neuropsychiatric manifestations such as optic neuritis, aseptic meningitis, demyelinating disease, etc. Lifelong warfarin is indicated in cases of thromboembolic CNS events associated with antiphospholipid antibody syndrome. High-dose corticosteroids can be used in cognitive impairment, although there is no robust data on this.
Renal manifestations: Lupus nephritis (LN) shall be confirmed with a biopsy, which confirms the diagnosis, rules out other causes and helps to classify the disease. Class I and II LN shall be treated with the Renin-angiotensin-aldosterone system blockade. Immunosuppression with high-dose corticosteroids followed by azathioprine is indicated only if proteinuria is more than 1 gram/day. Membranous LN (class V) shall also be treated with the Renin-angiotensin-aldosterone system blockade. If proteinuria of more than 1 gram/day is present (which is frequent in Class V LN), induction therapy with high dose corticosteroids and azathioprine (mild disease) or tacrolimus/cyclosporine-A/mycophenolate mofetil/IV cyclophosphamide (moderate to severe disease) followed by maintenance therapy with azathioprine, mycophenolate mofetil, cyclosporine-A or tacrolimus shall be used.
Corticosteroids shall be gradually tapered during maintenance therapy. Proliferative LN (class III/IV) requires more aggressive therapy. Induction therapy is with IV pulse dose methylprednisolone followed by high dose oral steroids combined with mycophenolate mofetil, IV cyclophosphamide, or azathioprine (only in mild disease in whites). Maintenance therapy with mycophenolate mofetil or azathioprine shall be continued for at least three years. IV pulse cyclophosphamide for one year can be considered maintenance therapy for severe disease. Lupus nephritis patients need very close monitoring of their renal function and proteinuria in addition to other SLE disease activity markers. Flares and incomplete remission are common. Renal replacement therapy and transplant may be needed in some patients.[6]
Pregnancy manifestations: Pregnancy shall be considered only if the disease was quiescent at the time and six months prior due to an increased risk of flares otherwise. Contraception, if needed, shall be used until then and shall be progesterone-only contraception. Hydroxychloroquine is considered safe in pregnancy, has been associated with a significant reduction in flares and disease activity, and shall be continued through pregnancy. Azathioprine and low-dose corticosteroids can be used for mild manifestations. Other immunosuppressive agents, including methotrexate, leflunomide, mycophenolate mofetil, cyclophosphamide, are teratogenic and contraindicated in pregnancy.
Rituximab and belimumab shall also be avoided during pregnancy. Patients with antiphospholipid antibody syndrome shall transition from warfarin to low-molecular-weight heparin and aspirin before pregnancy. For females with positive Anti-Ro or Anti-La antibodies with a history of neonatal lupus in a previous pregnancy, close fetal heart monitoring with weekly or alternate-weekly fetal echocardiography is recommended during the second trimester. First or second-degree heart block shall be promptly treated with dexamethasone, although prophylaxis with dexamethasone is not recommended. A complete heart block is irreversible and requires a permanent pacemaker in the infant. Hydroxychloroquine decreases the risk of fetal heart block.
Other management considerations: Hydroxychloroquine shall be used in all patients with SLE given its benefits beyond just managing active manifestations, including anti-thrombotic properties, and preventing flares. Patients on hydroxychloroquine will require regular ophthalmology exams to monitor for the rare but irreversible maculopathy associated with this drug. Corticosteroids are frequently used in SLE, with many patients unable to taper them completely. Long-term adverse effects of corticosteroids shall be considered and monitored, including osteoporosis, glaucoma, cataract, and avascular necrosis. Patients on high-dose corticosteroids will also need antibiotic prophylaxis to prevent infections. Most immunosuppressive agents used in SLE have several potential adverse effects ranging from cytopenias and hepatotoxicity with most to an increased risk of urinary bladder cancer with cyclophosphamide. These patients shall be appropriately and closely monitored for adverse effects of these agents.
Differential Diagnosis
SLE is a systemic disease with multiorgan involvement, and several other diseases can mimic SLE.
- Other Autoimmune Diseases
- Rheumatoid arthritis (RA) can present with several extra-articular manifestations in addition to the classic polyarticular inflammatory arthritis and may be difficult to differentiate from SLE. Positive ANA, Anti-Ro, and Anti-La can also be seen in RA, although other SLE-specific autoantibodies and hypocomplementemia are rare. SLE can be associated with a positive rheumatoid factor, but the Anti-CCP is negative in SLE
- Drug-induced lupus may be difficult to differentiate from SLE, especially due to a significant overlap in the clinical and serological features. Drug-induced lupus is characterized by the resolution of symptoms after drug withdrawal and lack of more severe manifestations, although the autoantibodies may remain positive for several years.
- Adult-onset Still disease characterized by arthralgia, fever, lymphadenopathy, and splenomegaly but no malar rash or other organ manifestations and lacks the SLE specific autoantibodies.
- Behcet disease presents with aphthous ulcers, uveitis, and arthralgia but lacks the other systemic and serological features of SLE.
- Sarcoidosis presents with fever, cough, dyspnea, fatigue, night sweats, rash, and uveitis. It shows non-caseating granuloma on chest radiography and bilateral adenopathy, which is rarely present in SLE.[23]
- Infections
- Several viral infections can mimic SLE. Parvovirus B19 infection can cause fever, rash, inflammatory arthritis, and cytopenias. ANA and rheumatoid factor have been reported. Hepatitis B and C can be associated with arthralgia/inflammatory arthritis and positive ANA and rheumatoid factor. CMV and EBV viral infections can cause fever, fatigue, cytopenias, and transaminitis. HIV can cause fever, fatigue, oral ulcers, and cytopenias. More specific autoantibodies and systemic manifestations of SLE are absent in these viral infections. Further, positive viral serologies may help make the right diagnosis.
- Infectious endocarditis characterized by fever, arterial emboli, arthralgia, myalgia, and a heart murmur; may be confused with cardiac manifestations of SLE but can be differentiated by the absence of specific SLE associated autoantibodies and positive blood cultures.[22]
- Malignancies
- Lymphomas, especially non-Hodgkins lymphoma, can present with fatigue, weight loss, fever, arthralgia, cytopenia, lymphadenopathy, and a positive ANA. The more specific SLE-associated autoantibodies are absent. In elderly patients presenting with lupus-like symptoms, malignancy shall be ruled out by cancer screening.
Prognosis
Despite the advancements in therapeutic options of SLE and a better understanding of the disease process, SLE patients suffer from significant morbidity and carry a high mortality. Survival rates are 85 to 90% during the first ten years. Leading causes of mortality include cardiovascular disease, infections, and renal disease. Early diagnosis with therapy to prevent organ damage, monitoring and screening patients for cardiovascular disease and infections with early intervention may improve these outcomes.
Poor prognostic factors in SLE include African American ethnicity, renal disease (especially diffuse proliferative glomerulonephritis), male sex, young age, older age at presentation, hypertension, low socioeconomic status, antiphospholipid antibody syndrome, presence of antiphospholipid antibodies, and high overall disease activity.[24]
Complications
Complications in patients with SLE may occur either due to organ damage by the disease or due to the adverse effects of the medications.
Disease process-related complications include but are not limited to accelerated atherosclerosis with a several-fold higher risk of coronary artery disease even in the younger population, end-stage renal disease, and neurological deficits, including blindness secondary to neuropsychiatric manifestations. Patients with severe cutaneous lupus, especially discoid lupus, can suffer permanent skin damage and alopecia. Anxiety and depression are more common in patients with SLE. Several pregnancy-related complications are well known, including fetal loss, pre-eclampsia and eclampsia, congenital heart block, and neonatal lupus.
Medication-induced complications are common and require close monitoring. Long-term corticosteroid use in SLE patients frequently leads to under-diagnosed and under-treated osteoporosis, leading to osteoporotic fractures. Other complications of long-term use of corticosteroid therapy include avascular necrosis, glaucoma, cataract, weight gain, and poor control of Diabetes mellitus. High-dose corticosteroid use can also be associated with opportunistic infections and acute psychosis. Long-term use of hydroxychloroquine may rarely result in maculopathy and retinopathy that is irreversible, and close ophthalmology examinations are recommended. Cyclophosphamide use is associated with a significantly high risk of interstitial cystitis and bladder cancer even after drug discontinuation. SLE patients are immunocompromised and at a significantly high risk of infections which is one of the significant causes of morbidity and mortality in SLE.[15]
Deterrence and Patient Education
As noted above, patient education is vital in the management of SLE. Better education of patients about the potential signs and symptoms of organ involvement may lead to early recognition and intervention, which may prevent organ damage. Further, patients shall be educated about the importance of compliance with medications. Dietary and lifestyle modification, including photoprotection and smoking cessation, is important. Patients with SLE suffer from significant stress related to the disease and complications and have higher rates of anxiety and depression. Support groups, behavioral therapy, and sometimes the involvement of psychiatry may help.
Enhancing Healthcare Team Outcomes
Lupus is a chronic inflammatory disorder with no cure. It can affect many organs and leads to poor quality of life without appropriate management. Premature death is common for a variety of causes. To reduce morbidity and mortality, an interprofessional team should educate and manage patients with SLE.
The primary care provider and nurse practitioner should educate the patient on avoiding triggers that cause flare-ups. In addition, the patient should be told to avoid UV light and minimize exposure to the sun. Appropriate garments, sunglasses, and a wide brim hat are recommended when going out. The dietitian should educate the patient on the importance of a low-fat diet to prevent hyperlipidemia. In addition, because patients with lupus are told to avoid the sun, vitamin D supplements are recommended. The physical therapist should educate the patient on the importance of exercise. The pharmacist should educate the patient on the importance of medication compliance and avoiding smoking. The nurse practitioner should counsel the patient on family planning and contraception. Many drugs used to treat are teratogenic, and thus, contraception is highly recommended. Multispeciality involvement is often needed in managing and monitoring SLE. While the involvement of a rheumatologist is crucial, other specialties including dermatology, cardiology, neurology, pulmonology, ophthalmology, nephrology, gastroenterology, and gynecology may be needed. Close communication between the providers and the patient and family and consideration of patient preferences while deciding therapy is strongly recommended. [Level 5]