Continuing Education Activity
Hypoaldosteronism is a clinical condition characterized by a deficiency of aldosterone or its impaired action at the tissue level. The disorder may result from disturbances in renal renin production and secretion, conversion of angiotensin I to angiotensin II, adrenal aldosterone synthesis and secretion, or from abnormal responsiveness of the target tissues to aldosterone. This activity outlines the evaluation and management of hypoaldosteronism and highlights the role of the interprofessional team in improving patient outcomes.
Objectives:
- Identify the etiology of hypoaldosteronism in both children and adults.
- Describe the pathophysiology of hypoaldosteronism in various age groups.
- Summarize the management of hypoaldosteronism in infants, children, and older adults.
- Explain the importance of collaboration and communication among the interprofessional team to ensure appropriate testing and treatment strategies for hypoaldosteronism to improve patient outcomes.
Introduction
Hypoaldosteronism (HA) is a condition marked by decreased synthesis or diminished release of aldosterone (ALD) from the zona glomerulosa of the adrenal glands, or resistance to its action on target tissues. In conditions of resistance, aldosterone levels are often elevated and termed pseudo-hypoaldosteronism.
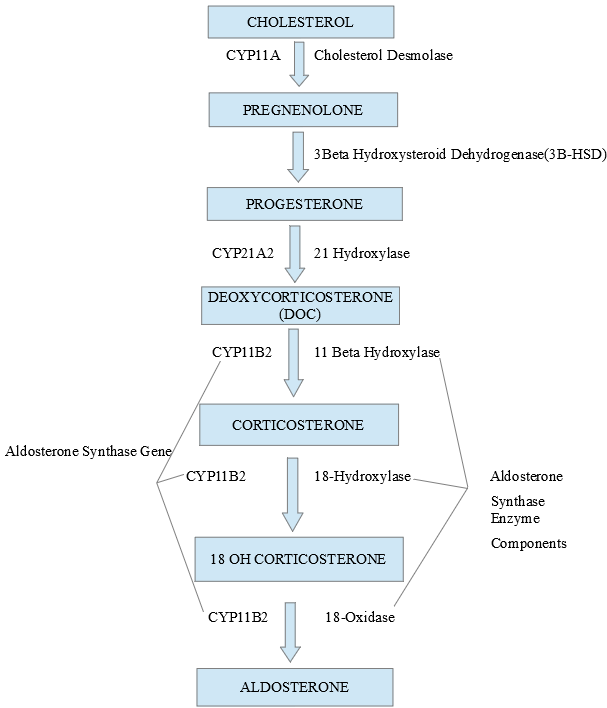
Recent advances have unraveled the mechanisms involved in the synthesis, release, and action of aldosterone on target organs. It is important first to understand these concepts to comprehend abnormalities in related diseases. The zona glomerulosa (ZG), which is the outermost layer of the adrenal cortex, is unique in possessing the key gene (CYP11B2) and enzyme (aldosterone synthase) for ALD synthesis which are absent from the other layers of the adrenal cortex. Likewise, the ZG is deficient in the machinery (CYP17 gene and related enzymes) for cortisol synthesis (see image 2).
ACTH and other neuropeptides play a less important role, while potassium (K+) and angiotensin II (Ang II) are the principal regulators of aldosterone. K+ regulates ALD independent of Ang II. The renin-angiotensin-aldosterone (RAA) axis is a feedback system that tightly regulates sodium (Na), K+, water, extracellular compartment fluid (ECF) volume, and blood pressure. A drop in perfusion will trigger the cells of the macula densa of the juxtaglomerular apparatus to secrete renin, which cleaves the hepatocyte derived protein angiotensinogen to angiotensin I (Ang I). Angiotensin-converting enzyme (ACE) in the vascular endothelium converts Ang I to Ang II, which is the most potent stimulus for aldosterone production and release.
Amiloride sensitive sodium channels are located in the distal renal tubular and collecting duct epithelial cells (ENaC). They are composed of three subunits alfa, beta, and gamma. Aldosterone mediates both genomic (ENaC gene transcription) and non-genomic (decreased ENaC degradation and hence enhanced surface expression of ENaC) effects through the mineralocorticoid receptor (MR) belonging to the family of intracellular nuclear receptors. The ENaC facilitates passive energy independent Na reabsorption. Apart from the kidneys, MR is present in the epithelia of the distal colon, sweat glands, salivary glands, airways, eyes, and nonepithelial cardiovascular and central nervous tissues.[1]
Both cortisol and aldosterone have an equal affinity to the MR. Although cortisol levels are much higher, the 11beta HSD2 in renal tubules converts active cortisol to inactive cortisone, thereby allowing aldosterone to dominate receptor binding.[2][3][4]
Etiology
Isolated Aldosterone Defects
A) Defective synthesis: Hyperreninemic hypoaldosteronism (renin-angiotensin independent; renin production is intact)
Immaturity
Immaturity of aldosterone synthetic enzymes in VPT infants
Aldosterone synthase deficiency I and II
Critically ill patients
Delayed recovery of the suppressed gland after contralateral adrenalectomy
Drug-induced diminished synthesis
Discontinuation of drugs with mineralocorticoid activity after prolonged use
B) Defective release:
Hyporeninemic hypoaldosteronism (renin-angiotensin dependent; decreased renin or Ang II production results in decreased aldosterone production
a) Congenital.
b) Acquired (type 4 RTA).
-
Diabetes
-
Various nephropathies
-
Autonomic neuropathy
-
Sickle cell disease
-
HIV disease
C) Medications such as NSAIDs, COX-2 inhibitors, ACEI/ARBs
D) Aldosterone resistance:
Genetic
i) Autosomal dominant pseudohypoaldosteronism (PHA AD)
ii) Autosomal recessive pseudohypoaldosteronism (PHA AR)
Acquired
i) Urinary tract infection (UTI) and obstructive uropathy (PHA III, resistance to ALD)[5]
ii) Medications
iii) Downregulation of MR in renal tubules (solid organ transplant)
Adrenal failure due to congenital or acquired causes can sometimes present initially as isolated aldosterone deficiency and later on progresses to involve the cortisol axis.[6]
Hypoaldosteronism can occur in both congenital and acquired conditions as part of a general adrenal failure as in congenital adrenal hyperplasia (CAH), adrenal hypoplasia (AHC), or acquired adrenal destruction and are not discussed here.
Epidemiology
The prevalence of congenital causes of selective HA is low, and that of PHA AD type I is less than 1:80000. Some causes of genetically inherited PHA I may be found in higher frequencies in certain ethnic populations.
Acquired causes are more frequent and commoner in hospitalized patients, children with type1 diabetes or sickle cell disease, adults with type 1 and 2 diabetes, and older adults on multiple medications known to affect the RAA axis. The incidence of contralateral adrenal suppression after adrenalectomy for Conn syndrome is estimated at around 5 percent.
Hyperkalemia occurs with increased frequency in recipients of solid organ transplantation and subsequent use of calcineurin inhibitors and is estimated between 40 and 70 percent.[7]
Pathophysiology
In prematurity, the delayed maturation of the adrenal synthetic pathways despite appropriate renin-angiotensin stimulation results in hypoaldosteronism that is most marked in infants with VPT. This is due to decreased activity of (CYP11B1) 11 beta-hydroxylase, which improves by day 3.[8]
Hypoaldosteronism may result from defective secretion of renin, angiotensin generation, or aldosterone synthesis or release.
Where the primary defect is in aldosterone synthesis, there is a compensatory increase in renin and Ang II causing hyperreninemic hypoaldosteronism as seen in aldosterone synthase defects (CYP11B2), chronic suppression of ALD secretion from the normal contralateral gland after unilateral adrenalectomy for Conn syndrome, and discontinuation of substances having mineralocorticoid activity after prolonged use. An example is licorice used in chewing tobacco, mouth fresheners, and herbal remedies.[9]
In critically ill patients, prolonged ACTH stimulation, along with cytokines released as a result of the critical illness, decreases the activity of aldosterone synthase and causes hyperreninemic HA and hypercortisolemia.
In hyporeninemic hypoaldosteronism or type 4 RTA, there is a decreased renin output and consequent hypoaldosteronism. Renin production is dependent on sympathetic stimulation, and defective renin release results from autonomic neuropathies. This is aggravated by concomitant renal disease, medications affecting the RAA axis, the production of inactive renin, a decreased beta-receptor sensitivity, and defective prostaglandin synthesis. Typical patients are in the sixth to the eighth decades of life, have diabetes, mild to moderate renal disease and are taking any medications affecting the RAA, and present with asymptomatic hyperkalemia, hyponatremia, and hyperchloremic normal anion gap acidosis. The serum ALD is low. Similarly, in patients with HIV, viral adrenalitis, nephropathy, and trimethoprim are contributory.
Beta-blockers decrease renin production, and NSAIDs inhibit cyclooxygenase and prevent renin release.
ACE inhibitors decrease Ang II production. ARBs block the stimulatory action of Ang II and prevent ALD secretion.
Spironolactone inhibits ALD biosynthesis as well as competitively inhibits the MR receptor. Amiloride and trimethoprim block the ENaC in the renal tubules increasing Na excretion and retaining K+. Triamterene is a K+ sparing diuretic acting on ALD independent ion-exchange sites in the DCT.
Both conventional and low molecular weight heparin (LMWH) decrease ALD production and release, and so do cyclosporin and calcium channel blockers. LMWH diminishes the sensitivity of the ZG to Ang II.
Drugs like ketoconazole used in Cushing disease and other cancers affect all three axes of the adrenal synthetic pathways and diminish the action of Ang II.
Dopamine tonically inhibits ALD production. Increased dopamine used in critical care and during therapy with dopamine agonists can, in certain situations, cause HA.
Autosomal dominant PHA is caused by mutations in the genes encoding the mineralocorticoid receptor (MR). Over fifty mutations have been identified, and in each of them, a single defective allele in either parent is enough to manifest the condition phenotypically.
Autosomal recessive PHA has loss of function mutations of the genes encoding the alfa, beta, and gamma subunits of the aldosterone-responsive, amiloride-sensitive ENaC, the genes that regulate posttranslational phosphorylation of the thiazide-sensitive sodium chloride cotransporter, and those that regulate phosphorylation and ubiquitination of cofactors that affect ENaC degradation.
Urinary tract infections and obstructive uropathy, especially in children, can result in temporary aldosterone resistance (PHA III).[5]
Patients post solid organ transplantation treated with calcineurin inhibitors cyclosporin and tacrolimus exhibit decreased MR transcription and down-regulation of MR receptors and consequent HA.[10]
History and Physical
Children with PHA AD have milder disease confined to the kidneys and may improve as they get older, whereas, those with PHA AR have more severe disease extending to involve the sweat and salivary glands, have frequent respiratory infections, skin rashes, ocular manifestations like retained meibomian gland secretions, and respiratory distress in the neonatal period.[11] Polyhydramnios may be a clue in the prenatal investigations to the presence of PHA AR. These children do not get better as they age and may require frequent hospitalizations and complex care.
PHA III patients will be cured once their UTI or obstructive uropathy resolves.
Rare syndromes with congenital abnormalities may have HA as a component.[12][13][14][15]
In adult patients, a good history will reveal the offending medications causing HA. Rare instances of life-threatening arrhythmias due to hyperkalemia have been reported in patients treated with conventional heparin.
A high index of suspicion should be maintained in hospitalized, the critically ill, those with diabetes, nephropathies from various causes, sickle cell disease, and HIV to facilitate early recognition and intervention of HA.
Patients who undergo adrenalectomy for unilateral ALD producing adenoma(Conn's syndrome) can have prolonged ZG suppression of the normal gland. Risk factors include advanced age, long-standing hypertension, large adenoma size, preoperative and postoperative renal function, and preoperative usage of NSAID.[16]
Evaluation
The key to an early accurate diagnosis in infants presenting with a salt-wasting crisis is to obtain a blood sample before therapy is begun. This is termed the 'critical sample,' and tests done on this specimen will avoid confounders from therapy. One aliquot is sent for analysis of serum electrolytes, aldosterone, 17 hydroxyprogesterone, and plasma renin activity or concentration. This will serve as the initial panel and will guide further testing.
A second aliquot is saved for confirmatory tests, which could include serum cortisol and ACTH in CAH, and 18 OHC and urine THA which are elevated and decreased respectively in type 1 as opposed to the type 2 ALD synthase deficiency. In infants with ALD synthase deficiency but normal aldosterone concentrations, a PAC/PRC ratio is useful to clarify the diagnosis of primary hypoaldosteronism.[17]
In some infants with very high ALD levels as part of PHA may exhibit a phenomenon called the 'hook effect' inherent in certain assay methods. In these infants, the initial ALD profiling may be reported as low, misleading the clinician to diagnose ALD synthase deficiency, but a repeat assay with serial sample dilutions will reveal very high ALD levels confirming PHA.[18]
Biochemistry:
Serum electrolytes
Serum and urine aldosterone
Serum cortisol and plasma ACTH
Serum 17 hydroxyprogesterone (17 OHP)
18 hydroxycorticosterone (18OHC) (low to normal in type 1 and high in type 2 ALD synthase deficiency
Urinary tetrahydro-aldosterone (THA), the metabolite of ALD (decreased in type 1 and low normal or normal in type 2 ALD synthase deficiency)
Plasma renin activity(PRA) or plasma renin concentration (PRC) if available
PAC/PRC ratio
PAC/K+ ratio( in CKD, less than 10 ng/dl is suggestive of HA and greater than 10 ng/dl is suggestive of renal aldosterone resistance)[19]
Serum creatinine and estimated glomerular filtration rate (eGFR)
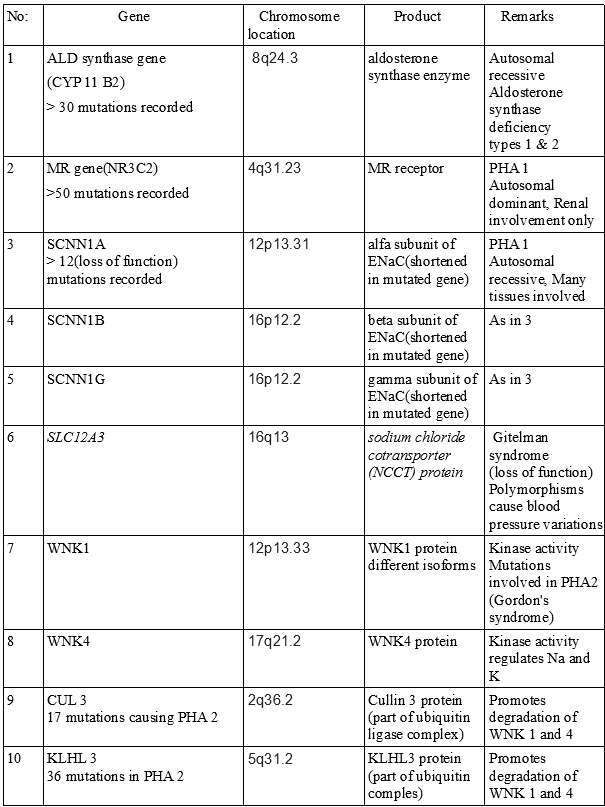
Molecular genetic studies including gene sequencing (see image 3)
Treatment / Management
The treatment of HA presenting in infancy and childhood with a salt-wasting crisis in ALD synthase deficiency and PHA AD is aggressive rehydration with NaCl. Before a definitive diagnosis of HA is made, after drawing a 'critical sample' of blood, the infants are treated for presumed CAH with intravenous hydrocortisone (HC) in stress doses. This also has adequate mineralocorticoid activity and will tide over the crises. If 17 OHP levels are in range, the HC can be discontinued after the infant stabilizes, and the mineralocorticoid fludrocortisone (FC) can be substituted orally at doses starting at 150 micrograms / m2 body surface area in ALD synthase deficiency. The younger the child, the greater are the doses of fludrocortisone because of drug insensitivity in infants and young children. Fludrocortisone is ineffective in PHA and NaCl, sodium bicarbonate, cation exchange resins, and in very severe cases, peritoneal dialysis is needed to counter hyperkalemia; thiazide diuretics to reduce hyperkalemia and hypercalciuria and indomethacin to counter polyuria, Na loss and reduce hypercalciuria are useful in PHA AD.
PHA AR is a much more severe disease with multi-organ involvement and may need very high doses of salt replacement and often show resistance to therapy. Carbenoxolone, an 11 beta HSD 2 inhibitor, prevents cortisol degradation and allows it to stimulate the MR receptor and function as a mineralocorticoid that may be useful in PHA AR.
In Conn syndrome, preoperatively, spironolactone, if used to control hypertension, should be stopped at least 3 days before surgery and any volume expansion corrected. Post adrenalectomy hypoaldosteronism is managed with fludrocortisone. Some patients may need extended support until the suppressed adrenal recovers.[16]
Renal transplant patients treated with calcineurin inhibitors present with hyperkalemia secondary to HA and respond to a low dose of fludrocortisone.[20]
In type 4 RTA, fludrocortisone is useful in reducing hyperkalemia, but caution is required as Na retention, fluid overload, and precipitation of congestive heart failure are side-effects. The addition of a loop diuretic or even better thiazides that block the NCCT in the distal renal tubules may be necessary. Precipitating medications like NSAID and K+ sparing diuretics must be carefully reviewed and discontinued.
Differential Diagnosis
Hypoaldosteronism can be differentiated from global adrenal failure by the presence of genital ambiguity in female infants and a high 17 OHP in both sexes in CAH. Addison's disease in older children and adults will have decreased serum cortisol and increased ACTH, present dramatically, and have physical findings like hyperpigmentation. Infants with congenital adrenal hypoplasia (AHC) present with salt-wasting and hyperpigmentation; cortisol deficiency appears later. It may be necessary to perform a cosyntropin stimulation test to confirm cortisol deficiency. Deficiency of cholesterol side-chain cleaving enzymes produce deficiencies in all three adrenal axes. Thus, salt-wasting, low cortisol, and ambiguous genitalia in male infants are seen.[21]
Decreased ALD and increased PRC is a feature of ALD synthase deficiency, whereas PHA has very high PRC and ALD, although both conditions have low Na, high K+, and acidosis.
In the two forms of ALD synthase deficiency, 18 OHC is low to normal in type 1 and high in type 2 ALD synthase deficiency, and the urinary tetrahydro-aldosterone (THA), the metabolite of ALD is decreased in type 1 and low normal or normal in type 2 ALD synthase deficiency.[17]
A definitive diagnosis of biosynthetic defects causing HA is by gene sequencing.[3]
A preoperative aldosterone contralateral suppression index (CSI -see image 1) during adrenal venous sampling (AVS) is the ratio of ALD to Cortisol in the ipsilateral adrenal vein divided by a similar ratio in the external iliac vein. Patients with a contralateral suppression index of <0.47 are at risk for prolonged adrenal suppression and require close monitoring of serum K+ after unilateral adrenalectomy for Conn syndrome.[22]
HA in the critically ill is not clinically significant due to high cortisol levels acting on the MR compensating for the absence of mineralocorticoid.
Some conditions have a low ALD level due to Na+ retention, hypertension, hypokalemia, suppressed PRC, and volume expansion. They should be distinguished from salt-losing causes of HA by the features mentioned. Examples are Liddle syndrome and the syndrome of mineralocorticoid excess due to 11 beta HSD2 inhibition. In Gordon syndrome, hypertension is accompanied by hyperkalemia, hyperchloremic acidosis, and suppressed renin due to volume expansion.
Prognosis
Prognosis is good in ALD synthase deficiency and PHA AD when identified early and promptly treated. Many children can be weaned off medications, and some go into remission as they grow. Older children who are inadequately treated have retarded growth or failure to thrive but adequately treated grow normally and have a good catch up growth. PHA AR patients do not as a rule improve and will need close monitoring. PHA III patients will be cured once their primary renal infection or obstruction is resolved.
Diligent management of medications causing HA will reduce morbidity considerably. Most patients with HA post-adrenalectomy for Conn syndrome resolve early, while some at high-risk need surveillance.
Complications
In patients treated for HA, overdosage with fludrocortisone can cause fluid retention, low K+, very suppressed PRC and, precipitate congestive heart failure. Less severe but clinically significant side-effects include constipation due to increased colonic mucosal Na and water reabsorption, weight gain due to fluid retention and muscle weakness due to hypokalemia. Dosing should be titrated to PRC levels in the upper limits of the normal range. Undertreated children will have poor growth. When the diagnosis is delayed in infants with severe disease, mortality is increased.
Consultations
- Endocrinology(pediatric and adult)
- Nephrology(pediatric and adult)
- Emergency medicine(pediatric and adult)
- Laboratory medicine
- Molecular genetics
- Clinical psychology
- Nursing
Deterrence and Patient Education
Mothers of children with congenital causes of HA should be educated in simple language about the nature of the disease, regularity in salt substitution and proper dosing of medication, symptoms, and signs of detecting overdosage and the prime importance of proper follow-up.
Conn's syndrome patients must receive proper education on the risk of hypoaldosteronism post-operatively. Patients with type 4 RTA will need education on avoiding over the counter NSAIDs to treat minor ailments and educated on their impact with their medical condition.
Pearls and Other Issues
Hypoaldosteronism in children is rare, but proper and timely recognition can be life-saving. Many advances have elucidated the mechanisms in the etiology, pathophysiology, genetics, evaluation, and management of HA, and a better understanding of these concepts will help reduce morbidity and mortality.
HA can occur isolated or as part of generalized adrenal failure.
In infancy, the presentation is often dramatic with a salt-wasting crisis; older children present with failure to thrive or poor growth. A high index of suspicion is required in children with hyperkalemia but a negative CAH screen. Hyperkalemia in the absence of renal insufficiency may indicate the presence of hypoaldosteronism.
ALD synthase deficiency is treated with NaCl and fludrocortisone and has a good prognosis, PHA AD and PHA AR are resistant to fludrocortisone, the former has a good prognosis while in the latter the outcome is poor due to generalized disease.
Identification of high-risk patients will aid better management of post-adrenalectomy complications in Conn syndrome.
The knowledge that various medications, including over the counter and alternative medicines cause HA, will improve patient outcomes.
High-risk patients with diabetes and mild renal dysfunction are prone to type 4 RTA. Care is required in diligently avoiding medications like potassium-sparing diuretics and NSAIDs and trimethoprim.
Enhancing Healthcare Team Outcomes
The initial presentation of the salt-wasting crisis is to the pediatric emergency physician. The general pediatrician takes over the infant's care after stabilization and provides follow-up with periodic input from the pediatric endocrinologist. A clinical geneticist may be necessary to confirm a diagnosis of causes of HA. Pediatric nephrologists are involved when intractable hyperkalemia needs dialysis therapy. Coordinated care by all specialties and effective communication among them will reduce mortality and morbidity, ensure the growth and development of these affected children. [Level 5] In every step, nursing and clinical psychology play important roles.
Adults with Conn syndrome will need coordinated care of the endocrinologist, endocrine surgeon, laboratory medicine personnel, interventional radiologist, and nurses skilled in the post-operative care to manage HA in these patients effectively.
Adults and older adults with type 4 RTA can be effectively managed by the primary care physician in coordination with other specialists and improve patient outcomes.