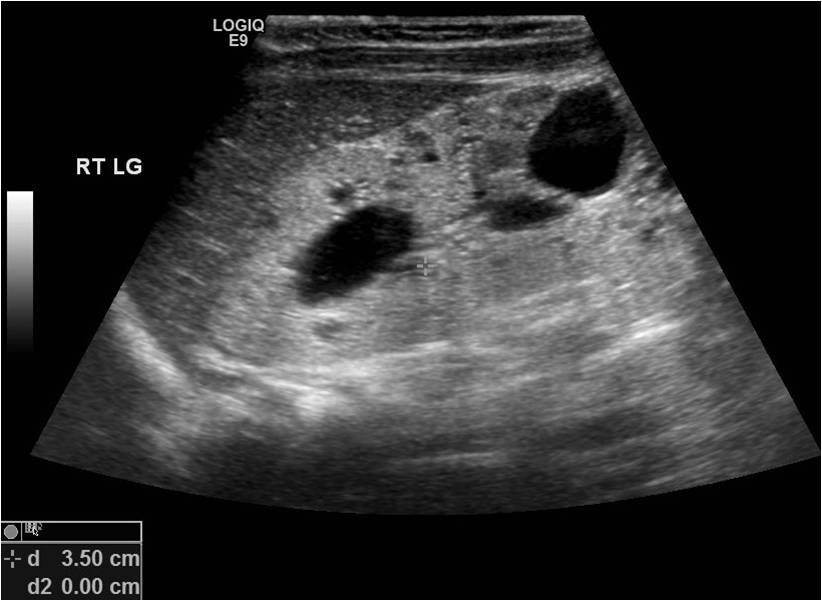
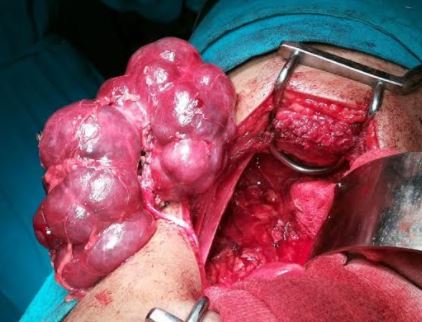
[2]
Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney international. 2009 Jul:76(2):149-68. doi: 10.1038/ki.2009.128. Epub 2009 May 20
[PubMed PMID: 19455193]
[3]
Mahboob M, Rout P, Bokhari SRA. Autosomal Dominant Polycystic Kidney Disease. StatPearls. 2024 Jan:():
[PubMed PMID: 30422529]
[4]
Guay-Woodford LM, Galliani CA, Musulman-Mroczek E, Spear GS, Guillot AP, Bernstein J. Diffuse renal cystic disease in children: morphologic and genetic correlations. Pediatric nephrology (Berlin, Germany). 1998 Apr:12(3):173-82
[PubMed PMID: 9630032]
[5]
Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nature reviews. Disease primers. 2018 Dec 6:4(1):50. doi: 10.1038/s41572-018-0047-y. Epub 2018 Dec 6
[PubMed PMID: 30523303]
[6]
Goggolidou P, Richards T. The genetics of Autosomal Recessive Polycystic Kidney Disease (ARPKD). Biochimica et biophysica acta. Molecular basis of disease. 2022 Apr 1:1868(4):166348. doi: 10.1016/j.bbadis.2022.166348. Epub 2022 Jan 12
[PubMed PMID: 35032595]
[7]
Liebau MC. Early clinical management of autosomal recessive polycystic kidney disease. Pediatric nephrology (Berlin, Germany). 2021 Nov:36(11):3561-3570. doi: 10.1007/s00467-021-04970-8. Epub 2021 Feb 17
[PubMed PMID: 33594464]
[8]
Guay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics. 2003 May:111(5 Pt 1):1072-80
[PubMed PMID: 12728091]
[9]
Bergmann C, Senderek J, Windelen E, Küpper F, Middeldorf I, Schneider F, Dornia C, Rudnik-Schöneborn S, Konrad M, Schmitt CP, Seeman T, Neuhaus TJ, Vester U, Kirfel J, Büttner R, Zerres K, APN (Arbeitsgemeinschaft für Pädiatrische Nephrologie). Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney international. 2005 Mar:67(3):829-48
[PubMed PMID: 15698423]
[10]
Alzarka B, Morizono H, Bollman JW, Kim D, Guay-Woodford LM. Design and Implementation of the Hepatorenal Fibrocystic Disease Core Center Clinical Database: A Centralized Resource for Characterizing Autosomal Recessive Polycystic Kidney Disease and Other Hepatorenal Fibrocystic Diseases. Frontiers in pediatrics. 2017:5():80. doi: 10.3389/fped.2017.00080. Epub 2017 Apr 20
[PubMed PMID: 28473971]
[11]
Cordido A, Vizoso-Gonzalez M, Garcia-Gonzalez MA. Molecular Pathophysiology of Autosomal Recessive Polycystic Kidney Disease. International journal of molecular sciences. 2021 Jun 17:22(12):. doi: 10.3390/ijms22126523. Epub 2021 Jun 17
[PubMed PMID: 34204582]
[12]
Guay-Woodford LM, Bissler JJ, Braun MC, Bockenhauer D, Cadnapaphornchai MA, Dell KM, Kerecuk L, Liebau MC, Alonso-Peclet MH, Shneider B, Emre S, Heller T, Kamath BM, Murray KF, Moise K, Eichenwald EE, Evans J, Keller RL, Wilkins-Haug L, Bergmann C, Gunay-Aygun M, Hooper SR, Hardy KK, Hartung EA, Streisand R, Perrone R, Moxey-Mims M. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. The Journal of pediatrics. 2014 Sep:165(3):611-7. doi: 10.1016/j.jpeds.2014.06.015. Epub 2014 Jul 9
[PubMed PMID: 25015577]
Level 3 (low-level) evidence
[13]
Bergmann C, Senderek J, Küpper F, Schneider F, Dornia C, Windelen E, Eggermann T, Rudnik-Schöneborn S, Kirfel J, Furu L, Onuchic LF, Rossetti S, Harris PC, Somlo S, Guay-Woodford L, Germino GG, Moser M, Büttner R, Zerres K. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Human mutation. 2004 May:23(5):453-63
[PubMed PMID: 15108277]
[14]
Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nature genetics. 2002 Mar:30(3):259-69
[PubMed PMID: 11919560]
[15]
Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, Rudnik-Schöneborn S, Mrug M, Sweeney W, Avner ED, Zerres K, Guay-Woodford LM, Somlo S, Germino GG. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. American journal of human genetics. 2002 May:70(5):1305-17
[PubMed PMID: 11898128]
[16]
Menezes LF, Cai Y, Nagasawa Y, Silva AM, Watkins ML, Da Silva AM, Somlo S, Guay-Woodford LM, Germino GG, Onuchic LF. Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney international. 2004 Oct:66(4):1345-55
[PubMed PMID: 15458427]
[17]
Traubici J, Daneman A. High-resolution renal sonography in children with autosomal recessive polycystic kidney disease. AJR. American journal of roentgenology. 2005 May:184(5):1630-3
[PubMed PMID: 15855129]
[18]
Bergmann C, Zerres K. Early manifestations of polycystic kidney disease. Lancet (London, England). 2007 Jun 30:369(9580):2157. doi: 10.1016/S0140-6736(07)61005-8. Epub
[PubMed PMID: 17604790]
[19]
Bergmann C. Genetics of Autosomal Recessive Polycystic Kidney Disease and Its Differential Diagnoses. Frontiers in pediatrics. 2017:5():221. doi: 10.3389/fped.2017.00221. Epub 2018 Feb 9
[PubMed PMID: 29479522]
[20]
Ward CJ, Yuan D, Masyuk TV, Wang X, Punyashthiti R, Whelan S, Bacallao R, Torra R, LaRusso NF, Torres VE, Harris PC. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Human molecular genetics. 2003 Oct 15:12(20):2703-10
[PubMed PMID: 12925574]
[22]
Richards WG, Sweeney WE, Yoder BK, Wilkinson JE, Woychik RP, Avner ED. Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease. The Journal of clinical investigation. 1998 Mar 1:101(5):935-9
[PubMed PMID: 9486961]
[23]
Martinez JR, Grantham JJ. Polycystic kidney disease: etiology, pathogenesis, and treatment. Disease-a-month : DM. 1995 Nov:41(11):693-765
[PubMed PMID: 7587886]
[24]
Hartung EA, Matheson M, Lande MB, Dell KM, Guay-Woodford LM, Gerson AC, Warady BA, Hooper SR, Furth SL. Neurocognition in children with autosomal recessive polycystic kidney disease in the CKiD cohort study. Pediatric nephrology (Berlin, Germany). 2014 Oct:29(10):1957-65. doi: 10.1007/s00467-014-2816-5. Epub 2014 May 15
[PubMed PMID: 24828609]
[25]
Shaikewitz ST, Chapman A. Autosomal recessive polycystic kidney disease: issues regarding the variability of clinical presentation. Journal of the American Society of Nephrology : JASN. 1993 Jun:3(12):1858-62
[PubMed PMID: 8338916]
[26]
Gunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman Gerstein M, Piwnica-Worms K, Choyke P, Daryanani KT, Turkbey B, Fischer R, Bernardini I, Sincan M, Zhao X, Sandler NG, Roque A, Douek DC, Graf J, Huizing M, Bryant JC, Mohan P, Gahl WA, Heller T. Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology. 2013 Jan:144(1):112-121.e2. doi: 10.1053/j.gastro.2012.09.056. Epub 2012 Oct 3
[PubMed PMID: 23041322]
[27]
Zerres K, Rudnik-Schöneborn S, Steinkamm C, Becker J, Mücher G. Autosomal recessive polycystic kidney disease. Journal of molecular medicine (Berlin, Germany). 1998 Apr:76(5):303-9
[PubMed PMID: 9587064]
[28]
Sweeney WE Jr, Avner ED. Pathophysiology of childhood polycystic kidney diseases: new insights into disease-specific therapy. Pediatric research. 2014 Jan:75(1-2):148-57. doi: 10.1038/pr.2013.191. Epub 2013 Oct 31
[PubMed PMID: 24336431]
[29]
Kaplan BS, Fay J, Shah V, Dillon MJ, Barratt TM. Autosomal recessive polycystic kidney disease. Pediatric nephrology (Berlin, Germany). 1989 Jan:3(1):43-9
[PubMed PMID: 2702087]
[30]
Gimpel C, Avni EF, Breysem L, Burgmaier K, Caroli A, Cetiner M, Haffner D, Hartung EA, Franke D, König J, Liebau MC, Mekahli D, Ong ACM, Pape L, Titieni A, Torra R, Winyard PJD, Schaefer F. Imaging of Kidney Cysts and Cystic Kidney Diseases in Children: An International Working Group Consensus Statement. Radiology. 2019 Mar:290(3):769-782. doi: 10.1148/radiol.2018181243. Epub 2019 Jan 1
[PubMed PMID: 30599104]
Level 3 (low-level) evidence
[31]
Turkbey B, Ocak I, Daryanani K, Font-Montgomery E, Lukose L, Bryant J, Tuchman M, Mohan P, Heller T, Gahl WA, Choyke PL, Gunay-Aygun M. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis (ARPKD/CHF). Pediatric radiology. 2009 Feb:39(2):100-11. doi: 10.1007/s00247-008-1064-x. Epub 2008 Dec 17
[PubMed PMID: 19089418]
[32]
Gleason DC, McAlister WH, Kissane J. Cystic disease of the kidneys in children. The American journal of roentgenology, radium therapy, and nuclear medicine. 1967 May:100(1):135-46
[PubMed PMID: 4960727]
[33]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Burgmaier K, Gimpel C, Schaefer F, Liebau M. Autosomal Recessive Polycystic Kidney Disease – PKHD1. GeneReviews(®). 1993:():
[PubMed PMID: 20301501]
[34]
Hoyer PF. Clinical manifestations of autosomal recessive polycystic kidney disease. Current opinion in pediatrics. 2015 Apr:27(2):186-92. doi: 10.1097/MOP.0000000000000196. Epub
[PubMed PMID: 25689455]
Level 3 (low-level) evidence
[35]
Mekahli D, Liebau MC, Cadnapaphornchai MA, Goldstein SL, Greenbaum LA, Litwin M, Seeman T, Schaefer F, Guay-Woodford LM. Design of two ongoing clinical trials of tolvaptan in the treatment of pediatric patients with autosomal recessive polycystic kidney disease. BMC nephrology. 2023 Feb 13:24(1):33. doi: 10.1186/s12882-023-03072-x. Epub 2023 Feb 13
[PubMed PMID: 36782137]
[36]
Edrees BM, Athar M, Al-Allaf FA, Taher MM, Khan W, Bouazzaoui A, Al-Harbi N, Safar R, Al-Edressi H, Alansary K, Anazi A, Altayeb N, Ahmed MA, Abduljaleel Z. Next-generation sequencing for molecular diagnosis of autosomal recessive polycystic kidney disease. Gene. 2016 Oct 10:591(1):214-226. doi: 10.1016/j.gene.2016.07.021. Epub 2016 Jul 9
[PubMed PMID: 27401137]
[37]
Melchionda S, Palladino T, Castellana S, Giordano M, Benetti E, De Bonis P, Zelante L, Bisceglia L. Expanding the mutation spectrum in 130 probands with ARPKD: identification of 62 novel PKHD1 mutations by sanger sequencing and MLPA analysis. Journal of human genetics. 2016 Sep:61(9):811-21. doi: 10.1038/jhg.2016.58. Epub 2016 May 26
[PubMed PMID: 27225849]
[38]
Yang H, Sieben CJ, Schauer RS, Harris PC. Genetic Spectrum of Polycystic Kidney and Liver Diseases and the Resulting Phenotypes. Advances in kidney disease and health. 2023 Sep:30(5):397-406. doi: 10.1053/j.akdh.2023.04.004. Epub
[PubMed PMID: 38097330]
Level 3 (low-level) evidence
[39]
Society for Maternal-Fetal Medicine (SMFM), Swanson K. Autosomal recessive polycystic kidney disease. American journal of obstetrics and gynecology. 2021 Nov:225(5):B7-B8. doi: 10.1016/j.ajog.2021.06.038. Epub 2021 Sep 8
[PubMed PMID: 34507795]
[40]
Wicher D, Obrycki Ł, Jankowska I. Autosomal Recessive Polycystic Kidney Disease-The Clinical Aspects and Diagnostic Challenges. Journal of pediatric genetics. 2021 Mar:10(1):1-8. doi: 10.1055/s-0040-1714701. Epub 2020 Jul 29
[PubMed PMID: 33552631]
[41]
Bergmann C, Senderek J, Schneider F, Dornia C, Küpper F, Eggermann T, Rudnik-Schöneborn S, Kirfel J, Moser M, Büttner R, Zerres K. PKHD1 mutations in families requesting prenatal diagnosis for autosomal recessive polycystic kidney disease (ARPKD). Human mutation. 2004 May:23(5):487-95
[PubMed PMID: 15108281]
[42]
Dukic L, Schaffelder R, Schaible T, Sütterlin M, Siemer J. [Massive increase of foetal abdominal circumference due to hereditary polycystic kidney disease]. Zeitschrift fur Geburtshilfe und Neonatologie. 2010 Jun:214(3):119-22. doi: 10.1055/s-0029-1225641. Epub 2010 Jun 23
[PubMed PMID: 20574939]
[43]
Belin S, Delco C, Parvex P, Hanquinet S, Fokstuen S, Martinez de Tejada B, Eperon I. Management of delivery of a fetus with autosomal recessive polycystic kidney disease: a case report of abdominal dystocia and review of the literature. Journal of medical case reports. 2019 Dec 12:13(1):366. doi: 10.1186/s13256-019-2293-3. Epub 2019 Dec 12
[PubMed PMID: 31829256]
Level 3 (low-level) evidence
[44]
Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics. 2014 Sep:134(3):e833-45. doi: 10.1542/peds.2013-3646. Epub 2014 Aug 11
[PubMed PMID: 25113295]
[45]
Lilova M, Kaplan BS, Meyers KE. Recombinant human growth hormone therapy in autosomal recessive polycystic kidney disease. Pediatric nephrology (Berlin, Germany). 2003 Jan:18(1):57-61
[PubMed PMID: 12488992]
[46]
Davis ID, Ho M, Hupertz V, Avner ED. Survival of childhood polycystic kidney disease following renal transplantation: the impact of advanced hepatobiliary disease. Pediatric transplantation. 2003 Oct:7(5):364-9
[PubMed PMID: 14738296]
[47]
Telega G, Cronin D, Avner ED. New approaches to the autosomal recessive polycystic kidney disease patient with dual kidney-liver complications. Pediatric transplantation. 2013 Jun:17(4):328-35. doi: 10.1111/petr.12076. Epub 2013 Apr 17
[PubMed PMID: 23593929]
[48]
Müller RU, Messchendorp AL, Birn H, Capasso G, Cornec-Le Gall E, Devuyst O, van Eerde A, Guirchoun P, Harris T, Hoorn EJ, Knoers NVAM, Korst U, Mekahli D, Le Meur Y, Nijenhuis T, Ong ACM, Sayer JA, Schaefer F, Servais A, Tesar V, Torra R, Walsh SB, Gansevoort RT. An update on the use of tolvaptan for autosomal dominant polycystic kidney disease: consensus statement on behalf of the ERA Working Group on Inherited Kidney Disorders, the European Rare Kidney Disease Reference Network and Polycystic Kidney Disease International. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2022 Apr 25:37(5):825-839. doi: 10.1093/ndt/gfab312. Epub
[PubMed PMID: 35134221]
Level 3 (low-level) evidence
[49]
Lacquaniti A. [Tolvaptan and autosomal dominant polycystic kidney disease in the adult: let's give time to the "TEMPO" trial (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes)]. Giornale italiano di nefrologia : organo ufficiale della Societa italiana di nefrologia. 2013 Jan-Feb:30(1):. pii: gin/30.1.10. Epub
[PubMed PMID: 25083527]
[50]
Torres VE, Higashihara E, Devuyst O, Chapman AB, Gansevoort RT, Grantham JJ, Perrone RD, Ouyang J, Blais JD, Czerwiec FS, TEMPO 3:4 Trial Investigators. Effect of Tolvaptan in Autosomal Dominant Polycystic Kidney Disease by CKD Stage: Results from the TEMPO 3:4 Trial. Clinical journal of the American Society of Nephrology : CJASN. 2016 May 6:11(5):803-811. doi: 10.2215/CJN.06300615. Epub 2016 Feb 23
[PubMed PMID: 26912543]
[51]
Chebib FT, Zhou X, Garbinsky D, Davenport E, Nunna S, Oberdhan D, Fernandes A. Tolvaptan and Kidney Function Decline in Older Individuals With Autosomal Dominant Polycystic Kidney Disease: A Pooled Analysis of Randomized Clinical Trials and Observational Studies. Kidney medicine. 2023 Jun:5(6):100639. doi: 10.1016/j.xkme.2023.100639. Epub 2023 Apr 14
[PubMed PMID: 37250503]
Level 1 (high-level) evidence
[54]
Van De Weghe JC, Gomez A, Doherty D. The Joubert-Meckel-Nephronophthisis Spectrum of Ciliopathies. Annual review of genomics and human genetics. 2022 Aug 31:23():301-329. doi: 10.1146/annurev-genom-121321-093528. Epub 2022 Jun 2
[PubMed PMID: 35655331]
[55]
Spahiu L, Behluli E, Grajçevci-Uka V, Liehr T, Temaj G. Joubert syndrome: Molecular basis and treatment. Journal of mother and child. 2022 Mar 1:26(1):118-123. doi: 10.34763/jmotherandchild.20222601.d-22-00034. Epub 2023 Feb 22
[PubMed PMID: 36803942]
[56]
Rodriguez MM. Congenital Anomalies of the Kidney and the Urinary Tract (CAKUT). Fetal and pediatric pathology. 2014 Oct-Dec:33(5-6):293-320. doi: 10.3109/15513815.2014.959678. Epub 2014 Oct 14
[PubMed PMID: 25313840]
[57]
Chen CP. Meckel syndrome: genetics, perinatal findings, and differential diagnosis. Taiwanese journal of obstetrics & gynecology. 2007 Mar:46(1):9-14
[PubMed PMID: 17389183]
[58]
Verhave JC, Bech AP, Wetzels JF, Nijenhuis T. Hepatocyte Nuclear Factor 1β-Associated Kidney Disease: More than Renal Cysts and Diabetes. Journal of the American Society of Nephrology : JASN. 2016 Feb:27(2):345-53. doi: 10.1681/ASN.2015050544. Epub 2015 Aug 28
[PubMed PMID: 26319241]
[59]
Salomon R, Saunier S, Niaudet P. Nephronophthisis. Pediatric nephrology (Berlin, Germany). 2009 Dec:24(12):2333-44. doi: 10.1007/s00467-008-0840-z. Epub 2008 Jul 8
[PubMed PMID: 18607645]
[63]
Park E, Lee JM, Ahn YH, Kang HG, Ha II, Lee JH, Park YS, Kim NK, Park WY, Cheong HI. Hepatorenal fibrocystic diseases in children. Pediatric nephrology (Berlin, Germany). 2016 Jan:31(1):113-9. doi: 10.1007/s00467-015-3185-4. Epub 2015 Aug 11
[PubMed PMID: 26260382]
[64]
Gunay-Aygun M. Liver and kidney disease in ciliopathies. American journal of medical genetics. Part C, Seminars in medical genetics. 2009 Nov 15:151C(4):296-306. doi: 10.1002/ajmg.c.30225. Epub
[PubMed PMID: 19876928]