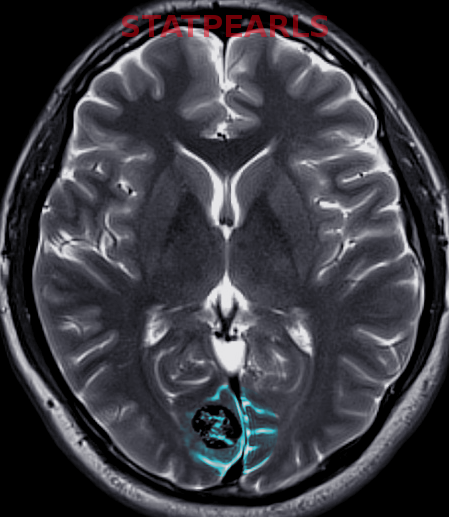
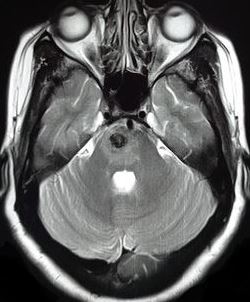
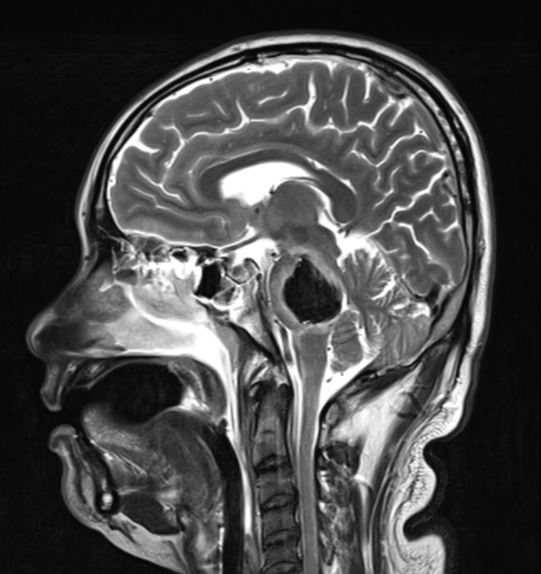
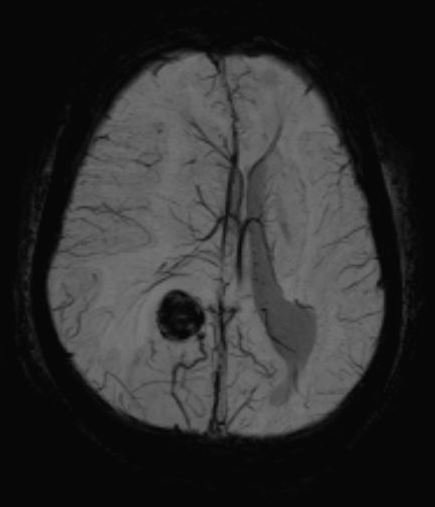
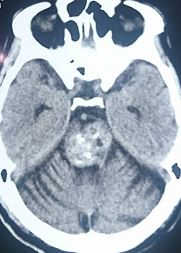
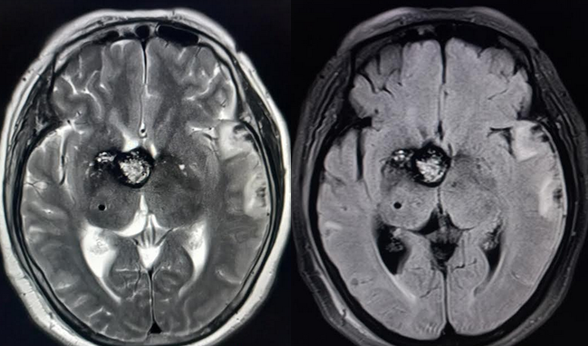
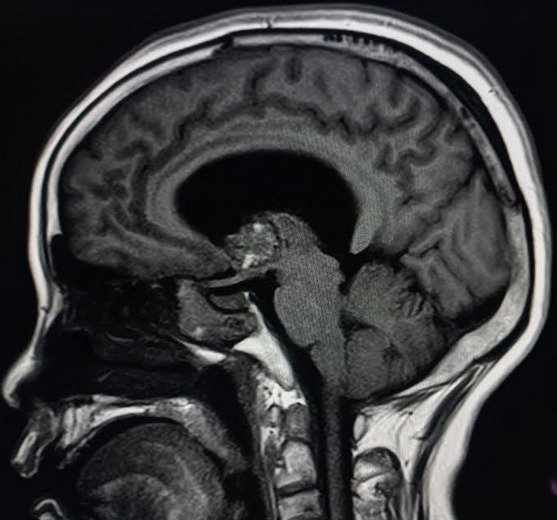
[1]
Rigamonti D, Hadley MN, Drayer BP, Johnson PC, Hoenig-Rigamonti K, Knight JT, Spetzler RF. Cerebral cavernous malformations. Incidence and familial occurrence. The New England journal of medicine. 1988 Aug 11:319(6):343-7
[PubMed PMID: 3393196]
[2]
Akers A, Al-Shahi Salman R, A Awad I, Dahlem K, Flemming K, Hart B, Kim H, Jusue-Torres I, Kondziolka D, Lee C, Morrison L, Rigamonti D, Rebeiz T, Tournier-Lasserve E, Waggoner D, Whitehead K. Synopsis of Guidelines for the Clinical Management of Cerebral Cavernous Malformations: Consensus Recommendations Based on Systematic Literature Review by the Angioma Alliance Scientific Advisory Board Clinical Experts Panel. Neurosurgery. 2017 May 1:80(5):665-680. doi: 10.1093/neuros/nyx091. Epub
[PubMed PMID: 28387823]
Level 3 (low-level) evidence
[3]
Otten P, Pizzolato GP, Rilliet B, Berney J. [131 cases of cavernous angioma (cavernomas) of the CNS, discovered by retrospective analysis of 24,535 autopsies]. Neuro-Chirurgie. 1989:35(2):82-3, 128-31
[PubMed PMID: 2674753]
Level 2 (mid-level) evidence
[4]
Flemming KD, Link MJ, Christianson TJ, Brown RD Jr. Prospective hemorrhage risk of intracerebral cavernous malformations. Neurology. 2012 Feb 28:78(9):632-6. doi: 10.1212/WNL.0b013e318248de9b. Epub 2012 Feb 1
[PubMed PMID: 22302553]
[5]
Al-Shahi Salman R, Berg MJ, Morrison L, Awad IA, Angioma Alliance Scientific Advisory Board. Hemorrhage from cavernous malformations of the brain: definition and reporting standards. Angioma Alliance Scientific Advisory Board. Stroke. 2008 Dec:39(12):3222-30. doi: 10.1161/STROKEAHA.108.515544. Epub 2008 Oct 30
[PubMed PMID: 18974380]
[6]
Labauge P, Denier C, Bergametti F, Tournier-Lasserve E. Genetics of cavernous angiomas. The Lancet. Neurology. 2007 Mar:6(3):237-44
[PubMed PMID: 17303530]
[7]
Jain R, Robertson PL, Gandhi D, Gujar SK, Muraszko KM, Gebarski S. Radiation-induced cavernomas of the brain. AJNR. American journal of neuroradiology. 2005 May:26(5):1158-62
[PubMed PMID: 15891176]
[8]
Fischer A, Zalvide J, Faurobert E, Albiges-Rizo C, Tournier-Lasserve E. Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends in molecular medicine. 2013 May:19(5):302-8. doi: 10.1016/j.molmed.2013.02.004. Epub 2013 Mar 15
[PubMed PMID: 23506982]
[9]
Zawistowski JS, Stalheim L, Uhlik MT, Abell AN, Ancrile BB, Johnson GL, Marchuk DA. CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Human molecular genetics. 2005 Sep 1:14(17):2521-31
[PubMed PMID: 16037064]
[10]
Greving JP, Wermer MJ, Brown RD Jr, Morita A, Juvela S, Yonekura M, Ishibashi T, Torner JC, Nakayama T, Rinkel GJ, Algra A. Development of the PHASES score for prediction of risk of rupture of intracranial aneurysms: a pooled analysis of six prospective cohort studies. The Lancet. Neurology. 2014 Jan:13(1):59-66. doi: 10.1016/S1474-4422(13)70263-1. Epub 2013 Nov 27
[PubMed PMID: 24290159]
[11]
Morris Z, Whiteley WN, Longstreth WT Jr, Weber F, Lee YC, Tsushima Y, Alphs H, Ladd SC, Warlow C, Wardlaw JM, Al-Shahi Salman R. Incidental findings on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ (Clinical research ed.). 2009 Aug 17:339():b3016. doi: 10.1136/bmj.b3016. Epub 2009 Aug 17
[PubMed PMID: 19687093]
Level 1 (high-level) evidence
[12]
Vernooij MW, Ikram MA, Tanghe HL, Vincent AJ, Hofman A, Krestin GP, Niessen WJ, Breteler MM, van der Lugt A. Incidental findings on brain MRI in the general population. The New England journal of medicine. 2007 Nov 1:357(18):1821-8
[PubMed PMID: 17978290]
[13]
Horne MA, Flemming KD, Su IC, Stapf C, Jeon JP, Li D, Maxwell SS, White P, Christianson TJ, Agid R, Cho WS, Oh CW, Wu Z, Zhang JT, Kim JE, Ter Brugge K, Willinsky R, Brown RD Jr, Murray GD, Al-Shahi Salman R, Cerebral Cavernous Malformations Individual Patient Data Meta-analysis Collaborators. Clinical course of untreated cerebral cavernous malformations: a meta-analysis of individual patient data. The Lancet. Neurology. 2016 Feb:15(2):166-173. doi: 10.1016/S1474-4422(15)00303-8. Epub 2015 Dec 2
[PubMed PMID: 26654287]
Level 1 (high-level) evidence
[14]
Del Curling O Jr, Kelly DL Jr, Elster AD, Craven TE. An analysis of the natural history of cavernous angiomas. Journal of neurosurgery. 1991 Nov:75(5):702-8
[PubMed PMID: 1919691]
[15]
Moore SA, Brown RD Jr, Christianson TJ, Flemming KD. Long-term natural history of incidentally discovered cavernous malformations in a single-center cohort. Journal of neurosurgery. 2014 May:120(5):1188-92. doi: 10.3171/2014.1.JNS131619. Epub 2014 Mar 14
[PubMed PMID: 24628608]
[16]
Ellis JA, Barrow DL. Supratentorial cavernous malformations. Handbook of clinical neurology. 2017:143():283-289. doi: 10.1016/B978-0-444-63640-9.00027-8. Epub
[PubMed PMID: 28552151]
[17]
Baumann CR, Schuknecht B, Lo Russo G, Cossu M, Citterio A, Andermann F, Siegel AM. Seizure outcome after resection of cavernous malformations is better when surrounding hemosiderin-stained brain also is removed. Epilepsia. 2006 Mar:47(3):563-6
[PubMed PMID: 16529622]
[18]
Al-Shahi Salman R, Hall JM, Horne MA, Moultrie F, Josephson CB, Bhattacharya JJ, Counsell CE, Murray GD, Papanastassiou V, Ritchie V, Roberts RC, Sellar RJ, Warlow CP, Scottish Audit of Intracranial Vascular Malformations (SAIVMs) collaborators. Untreated clinical course of cerebral cavernous malformations: a prospective, population-based cohort study. The Lancet. Neurology. 2012 Mar:11(3):217-24. doi: 10.1016/S1474-4422(12)70004-2. Epub 2012 Jan 31
[PubMed PMID: 22297119]
[19]
Washington CW, McCoy KE, Zipfel GJ. Update on the natural history of cavernous malformations and factors predicting aggressive clinical presentation. Neurosurgical focus. 2010 Sep:29(3):E7. doi: 10.3171/2010.5.FOCUS10149. Epub
[PubMed PMID: 20809765]
[20]
Rosenow F, Alonso-Vanegas MA, Baumgartner C, Blümcke I, Carreño M, Gizewski ER, Hamer HM, Knake S, Kahane P, Lüders HO, Mathern GW, Menzler K, Miller J, Otsuki T, Ozkara C, Pitkänen A, Roper SN, Sakamoto AC, Sure U, Walker MC, Steinhoff BJ, Surgical Task Force, Commission on Therapeutic Strategies of the ILAE. Cavernoma-related epilepsy: review and recommendations for management--report of the Surgical Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2013 Dec:54(12):2025-35. doi: 10.1111/epi.12402. Epub 2013 Oct 17
[PubMed PMID: 24134485]
[21]
Expert Panel on Neurologic Imaging:, Salmela MB, Mortazavi S, Jagadeesan BD, Broderick DF, Burns J, Deshmukh TK, Harvey HB, Hoang J, Hunt CH, Kennedy TA, Khalessi AA, Mack W, Patel ND, Perlmutter JS, Policeni B, Schroeder JW, Setzen G, Whitehead MT, Cornelius RS, Corey AS. ACR Appropriateness Criteria(®) Cerebrovascular Disease. Journal of the American College of Radiology : JACR. 2017 May:14(5S):S34-S61. doi: 10.1016/j.jacr.2017.01.051. Epub
[PubMed PMID: 28473091]
[22]
Gross BA, Lin N, Du R, Day AL. The natural history of intracranial cavernous malformations. Neurosurgical focus. 2011 Jun:30(6):E24. doi: 10.3171/2011.3.FOCUS1165. Epub
[PubMed PMID: 21631226]
[23]
Petersen TA, Morrison LA, Schrader RM, Hart BL. Familial versus sporadic cavernous malformations: differences in developmental venous anomaly association and lesion phenotype. AJNR. American journal of neuroradiology. 2010 Feb:31(2):377-82. doi: 10.3174/ajnr.A1822. Epub 2009 Oct 15
[PubMed PMID: 19833796]
[24]
Li D, Jiao YM, Wang L, Lin FX, Wu J, Tong XZ, Wang S, Cao Y. Surgical outcome of motor deficits and neurological status in brainstem cavernous malformations based on preoperative diffusion tensor imaging: a prospective randomized clinical trial. Journal of neurosurgery. 2018 Mar 16:130(1):286-301. doi: 10.3171/2017.8.JNS17854. Epub
[PubMed PMID: 29547081]
Level 1 (high-level) evidence
[25]
Pouratian N, Bookheimer SY, Rex DE, Martin NA, Toga AW. Utility of preoperative functional magnetic resonance imaging for identifying language cortices in patients with vascular malformations. Neurosurgical focus. 2002 Oct 15:13(4):e4
[PubMed PMID: 15771403]
[26]
Dammann P, Wrede K, Zhu Y, Matsushige T, Maderwald S, Umutlu L, Quick HH, Hehr U, Rath M, Ladd ME, Felbor U, Sure U. Correlation of the venous angioarchitecture of multiple cerebral cavernous malformations with familial or sporadic disease: a susceptibility-weighted imaging study with 7-Tesla MRI. Journal of neurosurgery. 2017 Feb:126(2):570-577. doi: 10.3171/2016.2.JNS152322. Epub 2016 May 6
[PubMed PMID: 27153162]
[27]
Moultrie F, Horne MA, Josephson CB, Hall JM, Counsell CE, Bhattacharya JJ, Papanastassiou V, Sellar RJ, Warlow CP, Murray GD, Al-Shahi Salman R, Scottish Audit of Intracranial Vascular Malformations (SAIVMs) steering committee and collaborators. Outcome after surgical or conservative management of cerebral cavernous malformations. Neurology. 2014 Aug 12:83(7):582-9. doi: 10.1212/WNL.0000000000000684. Epub 2014 Jul 3
[PubMed PMID: 24994841]
[28]
D'Angelo VA, De Bonis C, Amoroso R, Cali A, D'Agruma L, Guarnieri V, Muscarella LA, Zelante L, Bisceglia M, Scarabino T, Catapano D. Supratentorial cerebral cavernous malformations: clinical, surgical, and genetic involvement. Neurosurgical focus. 2006 Jul 15:21(1):e9
[PubMed PMID: 16859262]
[29]
Flores BC, Whittemore AR, Samson DS, Barnett SL. The utility of preoperative diffusion tensor imaging in the surgical management of brainstem cavernous malformations. Journal of neurosurgery. 2015 Mar:122(3):653-62. doi: 10.3171/2014.11.JNS13680. Epub 2015 Jan 9
[PubMed PMID: 25574568]
[30]
Abla AA, Lekovic GP, Turner JD, de Oliveira JG, Porter R, Spetzler RF. Advances in the treatment and outcome of brainstem cavernous malformation surgery: a single-center case series of 300 surgically treated patients. Neurosurgery. 2011 Feb:68(2):403-14; discussion 414-5. doi: 10.1227/NEU.0b013e3181ff9cde. Epub
[PubMed PMID: 21654575]
Level 2 (mid-level) evidence
[31]
Gross BA, Batjer HH, Awad IA, Bendok BR, Du R. Brainstem cavernous malformations: 1390 surgical cases from the literature. World neurosurgery. 2013 Jul-Aug:80(1-2):89-93. doi: 10.1016/j.wneu.2012.04.002. Epub 2012 Apr 5
[PubMed PMID: 22484766]
Level 3 (low-level) evidence
[32]
Atwal GS, Sarris CE, Spetzler RF. Brainstem and cerebellar cavernous malformations. Handbook of clinical neurology. 2017:143():291-295. doi: 10.1016/B978-0-444-63640-9.00028-X. Epub
[PubMed PMID: 28552152]
[33]
Yeon JY, Kim JS, Choi SJ, Seo DW, Hong SB, Hong SC. Supratentorial cavernous angiomas presenting with seizures: surgical outcomes in 60 consecutive patients. Seizure. 2009 Jan:18(1):14-20. doi: 10.1016/j.seizure.2008.05.010. Epub 2008 Jul 24
[PubMed PMID: 18656386]
[34]
Lu XY, Sun H, Xu JG, Li QY. Stereotactic radiosurgery of brainstem cavernous malformations: a systematic review and meta-analysis. Journal of neurosurgery. 2014 Apr:120(4):982-7. doi: 10.3171/2013.12.JNS13990. Epub 2014 Feb 7
[PubMed PMID: 24506243]
Level 1 (high-level) evidence
[35]
Hasegawa T, McInerney J, Kondziolka D, Lee JY, Flickinger JC, Lunsford LD. Long-term results after stereotactic radiosurgery for patients with cavernous malformations. Neurosurgery. 2002 Jun:50(6):1190-7; discussion 1197-8
[PubMed PMID: 12015835]
[36]
Nimjee SM, Powers CJ, Bulsara KR. Review of the literature on de novo formation of cavernous malformations of the central nervous system after radiation therapy . Neurosurgical focus. 2006 Jul 15:21(1):e4
[PubMed PMID: 16859257]
[37]
Shenkar R, Shi C, Austin C, Moore T, Lightle R, Cao Y, Zhang L, Wu M, Zeineddine HA, Girard R, McDonald DA, Rorrer A, Gallione C, Pytel P, Liao JK, Marchuk DA, Awad IA. RhoA Kinase Inhibition With Fasudil Versus Simvastatin in Murine Models of Cerebral Cavernous Malformations. Stroke. 2017 Jan:48(1):187-194. doi: 10.1161/STROKEAHA.116.015013. Epub 2016 Nov 22
[PubMed PMID: 27879448]
[38]
Hemphill JC 3rd, Greenberg SM, Anderson CS, Becker K, Bendok BR, Cushman M, Fung GL, Goldstein JN, Macdonald RL, Mitchell PH, Scott PA, Selim MH, Woo D, American Heart Association Stroke Council, Council on Cardiovascular and Stroke Nursing, Council on Clinical Cardiology. Guidelines for the Management of Spontaneous Intracerebral Hemorrhage: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2015 Jul:46(7):2032-60. doi: 10.1161/STR.0000000000000069. Epub 2015 May 28
[PubMed PMID: 26022637]
[39]
Schneble HM, Soumare A, Hervé D, Bresson D, Guichard JP, Riant F, Tournier-Lasserve E, Tzourio C, Chabriat H, Stapf C. Antithrombotic therapy and bleeding risk in a prospective cohort study of patients with cerebral cavernous malformations. Stroke. 2012 Dec:43(12):3196-9. doi: 10.1161/STROKEAHA.112.668533. Epub 2012 Nov 13
[PubMed PMID: 23150651]
[40]
McCracken DJ, Willie JT, Fernald BA, Saindane AM, Drane DL, Barrow DL, Gross RE. Magnetic Resonance Thermometry-Guided Stereotactic Laser Ablation of Cavernous Malformations in Drug-Resistant Epilepsy: Imaging and Clinical Results. Operative neurosurgery (Hagerstown, Md.). 2016 Mar:12(1):39-48. doi: 10.1227/NEU.0000000000001033. Epub 2015 Sep 25
[PubMed PMID: 27959970]
[41]
Zabramski JM, Wascher TM, Spetzler RF, Johnson B, Golfinos J, Drayer BP, Brown B, Rigamonti D, Brown G. The natural history of familial cavernous malformations: results of an ongoing study. Journal of neurosurgery. 1994 Mar:80(3):422-32
[PubMed PMID: 8113854]
[42]
Taslimi S, Modabbernia A, Amin-Hanjani S, Barker FG 2nd, Macdonald RL. Natural history of cavernous malformation: Systematic review and meta-analysis of 25 studies. Neurology. 2016 May 24:86(21):1984-91. doi: 10.1212/WNL.0000000000002701. Epub 2016 Apr 22
[PubMed PMID: 27164680]
Level 1 (high-level) evidence
[43]
Barker FG 2nd, Amin-Hanjani S, Butler WE, Lyons S, Ojemann RG, Chapman PH, Ogilvy CS. Temporal clustering of hemorrhages from untreated cavernous malformations of the central nervous system. Neurosurgery. 2001 Jul:49(1):15-24; discussion 24-5
[PubMed PMID: 11440436]
[44]
Kashefiolasl S, Bruder M, Brawanski N, Herrmann E, Seifert V, Tritt S, Konczalla J. A benchmark approach to hemorrhage risk management of cavernous malformations. Neurology. 2018 Mar 6:90(10):e856-e863. doi: 10.1212/WNL.0000000000005066. Epub 2018 Feb 2
[PubMed PMID: 29429974]
[45]
Porter PJ, Willinsky RA, Harper W, Wallace MC. Cerebral cavernous malformations: natural history and prognosis after clinical deterioration with or without hemorrhage. Journal of neurosurgery. 1997 Aug:87(2):190-7
[PubMed PMID: 9254081]
[46]
Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M, Maciazek J, Vicaut E, Brunereau L, Tournier-Lasserve E. Genotype-phenotype correlations in cerebral cavernous malformations patients. Annals of neurology. 2006 Nov:60(5):550-556. doi: 10.1002/ana.20947. Epub
[PubMed PMID: 17041941]
[47]
Rudy RF, Du R. Pharmacotherapy for cavernous malformations. Handbook of clinical neurology. 2017:143():309-316. doi: 10.1016/B978-0-444-63640-9.00031-X. Epub
[PubMed PMID: 28552155]
[48]
Englot DJ, Han SJ, Lawton MT, Chang EF. Predictors of seizure freedom in the surgical treatment of supratentorial cavernous malformations. Journal of neurosurgery. 2011 Dec:115(6):1169-74. doi: 10.3171/2011.7.JNS11536. Epub 2011 Aug 5
[PubMed PMID: 21819194]
[49]
Stavrou I, Baumgartner C, Frischer JM, Trattnig S, Knosp E. Long-term seizure control after resection of supratentorial cavernomas: a retrospective single-center study in 53 patients. Neurosurgery. 2008 Nov:63(5):888-96; discussion 897. doi: 10.1227/01.NEU.0000327881.72964.6E. Epub
[PubMed PMID: 19005379]
Level 2 (mid-level) evidence
[50]
Rathore C, Panda S, Sarma PS, Radhakrishnan K. How safe is it to withdraw antiepileptic drugs following successful surgery for mesial temporal lobe epilepsy? Epilepsia. 2011 Mar:52(3):627-35. doi: 10.1111/j.1528-1167.2010.02890.x. Epub 2011 Jan 10
[PubMed PMID: 21219315]
[51]
Schmidt D, Baumgartner C, Löscher W. Seizure recurrence after planned discontinuation of antiepileptic drugs in seizure-free patients after epilepsy surgery: a review of current clinical experience. Epilepsia. 2004 Feb:45(2):179-86
[PubMed PMID: 14738426]
[52]
Stockton RA, Shenkar R, Awad IA, Ginsberg MH. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. The Journal of experimental medicine. 2010 Apr 12:207(4):881-96. doi: 10.1084/jem.20091258. Epub 2010 Mar 22
[PubMed PMID: 20308363]
[53]
Borikova AL, Dibble CF, Sciaky N, Welch CM, Abell AN, Bencharit S, Johnson GL. Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype. The Journal of biological chemistry. 2010 Apr 16:285(16):11760-4. doi: 10.1074/jbc.C109.097220. Epub 2010 Feb 24
[PubMed PMID: 20181950]
[54]
McDonald DA, Shi C, Shenkar R, Stockton RA, Liu F, Ginsberg MH, Marchuk DA, Awad IA. Fasudil decreases lesion burden in a murine model of cerebral cavernous malformation disease. Stroke. 2012 Feb:43(2):571-4. doi: 10.1161/STROKEAHA.111.625467. Epub 2011 Oct 27
[PubMed PMID: 22034008]
[55]
Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, Ferrarini L, Orsenigo F, Papa E, Boulday G, Tournier-Lasserve E, Chapon F, Richichi C, Retta SF, Lampugnani MG, Dejana E. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature. 2013 Jun 27:498(7455):492-6. doi: 10.1038/nature12207. Epub 2013 Jun 9
[PubMed PMID: 23748444]
[56]
Bravi L, Rudini N, Cuttano R, Giampietro C, Maddaluno L, Ferrarini L, Adams RH, Corada M, Boulday G, Tournier-Lasserve E, Dejana E, Lampugnani MG. Sulindac metabolites decrease cerebrovascular malformations in CCM3-knockout mice. Proceedings of the National Academy of Sciences of the United States of America. 2015 Jul 7:112(27):8421-6. doi: 10.1073/pnas.1501352112. Epub 2015 Jun 24
[PubMed PMID: 26109568]