Continuing Education Activity
Canavan disease, also called Canavan-Van Bogaert-Bertrand disease, is a rare, progressive, and fatal neurological hereditary disorder that begins in infancy. It is part of a group of genetic diseases referred to as leukodystrophies. It is associated with the deficiency of an essential enzyme resulting in the loss of white matter in the brain, which subsequently leads to the defective transmission of nerve signals. This activity reviews the cause, presentation, pathophysiology and diagnosis of canavan disease and highlights the role of the interprofessional team in its management.
Objectives:
- Describe the pathophysiology of Canavan disease.
- Review the physical features of a patient with canavan disease.
- Summarize the treatment options for canavan disease.
- Explain the importance of improving care coordination among interprofessional team members to improve outcomes for patients affected by canavan disease.
Introduction
Canavan disease, also called Canavan-Van Bogaert-Bertrand disease, is a rare, progressive, and fatal neurological hereditary disorder that begins in infancy. It is part of a group of genetic diseases referred to as leukodystrophies. It is associated with the deficiency of an essential enzyme resulting in the loss of white matter in the brain, which subsequently leads to the defective transmission of nerve signals.[1][2]
The neonatal form is the most common and is usually associated with the most severe symptoms. Most infants appear healthy immediately after birth, but within 2 to 6 months, obvious deficits in gross motor development will be apparent. The infant may not be able to move, turn over, control head movements, or support itself. Hypotonia is a common feature and is usually associated with macrocephaly. Most infants have difficulty with feeding and develop seizures.[3][4]
Etiology
Canavan disease is an autosomal recessive disorder associated with a gene locus on chromosome 17. It is caused by a mutation in the ASPA gene that encodes the enzyme aspartoacylase. This mutation results in an aspartoacylase deficiency and the accumulation of N-acetylaspartic acid (NAA) in the brain. NAA is believed to cause oligodendrocyte dysfunction, consequential spongiform changes, and degeneration of myelin in the phospholipid layer of axons.[5]
Epidemiology
Canavan disease is one of the most prevalent degenerative cerebral diseases of infancy. It frequently affects Ashkenazi Jews but has been described in other populations as well. However, sufficient data is unavailable to calculate the prevalence. The carrier frequency among the Ashkenazi varies from 1:37 to 1:57 individuals, yielding approximate prevalence rates ranging from 1:6000 to 1:14,000.[6]
Pathophysiology
Acetyl-CoA and aspartic acid form NAA. It is believed to be the second most common free amino acid in the brain. It is found in neurons, where mitochondria synthesize it and then transfer it to oligodendrocytes through axo-glial contact zones. These axo-glial contact zones are formed between the innermost oligodendrocyte plasma membrane and the axonal membrane. NAA is converted to N-acetylaspartylglutamate (NAAG) within neurons. It is subsequently transported to astrocytes, which hydrolyze it to NAA and glutamate. The NAA is then finally taken up by oligodendrocytes which are the primary location of aspartoacylase.
The aspartoacylase enzyme catalyzes the conversion of NAA to aspartate (aspartic acid) and acetate. Therefore, deficiency of this catalyst leads to accumulation of NAA in the brain. Levels of NAA are markedly increased in the patient's plasma, urine, and cerebrospinal fluid. Abnormal myelination and the associated prominent swollen and vacuolated astrocytes are the fundamental hallmarks of aspartoacylase deficiency. These effects justify the name spongiform leukodystrophy. However, the precise mechanism by which accumulated NAA causes the pathogenesis of this spongiform degeneration remains uncertain.
History and Physical
Aspartoacylase deficiency initially presents generally around the age of three months. Typical features include lethargy, listlessness, weak cry and suck, a lack of head control when the baby is pulled from a lying to a sitting position, hypotonia, poor visual tracking or blindness, vomiting, and seizures. Some authors have also described poor feeding, irritability, and visual inattention.
Macrocephaly, or enlarged head size, becomes prominent by the age of three to six months. After that, hypotonia progresses to spasticity, hyperreflexia, extensor plantar responses, and tonic extensor spasms. The extensor spasms may occur in response to noise.
By the age of six months, neurologic irregularities are invariably present with little or no subsequent motor development.
Blindness due to optic atrophy occurs around the age of 6 to 18 months. Seizures, usually generalized tonic-clonic ones, are noted in about 50% of patients. Unlike most leukodystrophies, the cerebrospinal fluid protein is usually within the reference range.
Pseudobulbar signs and decerebrate posturing dominate the end stage of Canavan disease. Feeding is a major issue, with prominent swallowing dysfunction and gastroesophageal reflux.
Variant Forms
The presence of variant forms of aspartoacylase deficiency is controversial. A suggested juvenile-onset form, beginning after age five, is less common. Patients with this form of the disease will have slightly delayed speech and motor skill development since childhood. It is common for such delays to go under-recognized due to their mild and nonspecific nature. Onset in adults usually resembles multiple sclerosis on clinical grounds.
Evaluation
Laboratory Studies
In symptomatic infants with compatible clinical features and neuroimaging findings suggesting aspartoacylase deficiency, the diagnosis is concluded by observing elevated levels of urine NAA. Compatible clinical features include hypotonia, poor head control, and macrocephaly. A determination can be reached more specifically by detecting deficient aspartoacylase activity in cultured skin fibroblasts. If elevated urine NAA and skin fibroblast testing are diagnostic, genetic testing is obtained only for genetic counseling.[7][8][9]
Urine levels of NAA are increased up to 200 times that of the reference range. Gas chromatography or mass spectrometry are employed for measurement.
Prenatal Diagnosis
Prenatal diagnosis is best accomplished by measuring NAA levels in amniotic fluid. This method utilizes a stable isotope dilution coupled with gas chromatography or mass spectrometry, and also molecular analysis.
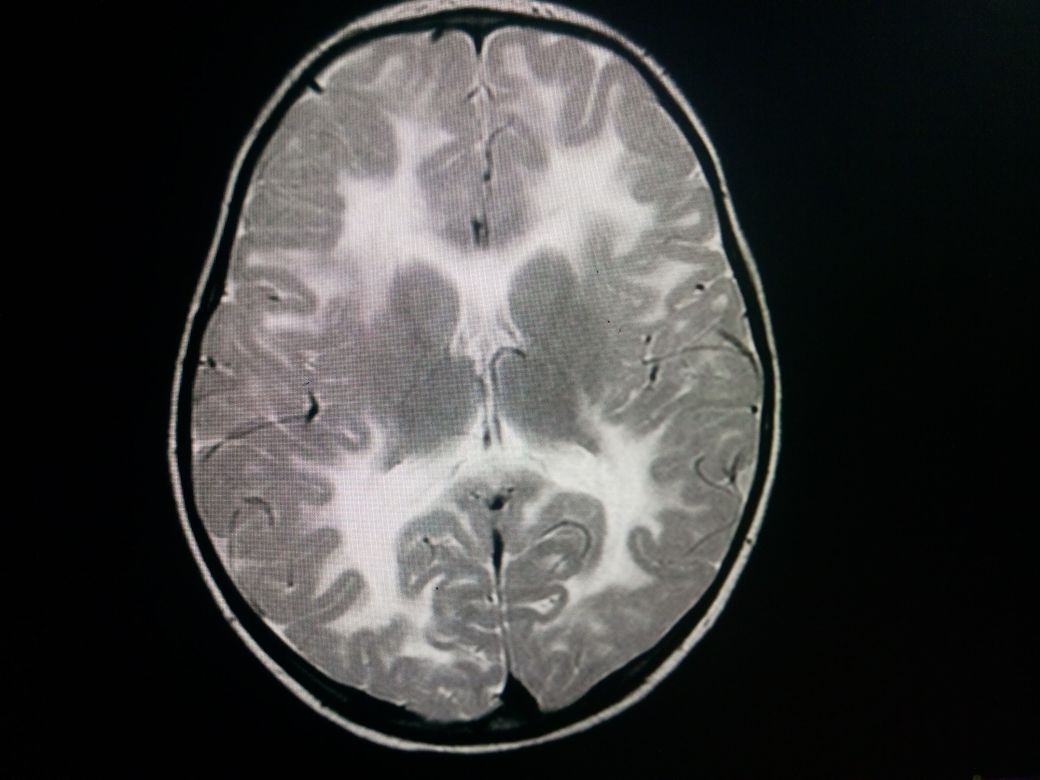
Neuroimaging
A CT scan shows a characteristic low attenuation of white matter in contrast to the relatively unaffected gray matter. This difference is mainly due to spongiform degeneration and edema of the white matter. No post contrast enhancement is usually seen.
MRI features include a relatively enlarged brain, also called megalencephaly. There is typically a diffuse bilateral white matter involvement. Unlike other leukodystrophies, subcortical U-fibers are usually involved early in the disease process. T1 shows a low signal in the white matter while T2 shows a high signal in white matter. There is no post contrast enhancement. MR spectroscopy shows markedly elevated levels of NAA, and an elevated NAA:creatine ratio. The mnemonic "CaNAAvan" is useful for remembering this information.
Treatment / Management
There is no currently available disease-modifying therapy for Canavan disease. Management aims to maintain nutrition and hydration, protect the airways, prevent seizures, minimize the risk of contractures, and cure infections. An assessment of the patient's nutritional and developmental status is recommended for guiding the management.
A feeding gastrostomy tube is typically used to maintain adequate nutrition and hydration in the presence of dysphagia. A gastrostomy tube can also decrease the risk of aspiration due to the patient's lack of sound reflexes.
Seizures are managed with standard antiepileptic drugs. Physical therapy and position changes are helpful to reduce the risk of contractures and ulcers. They also help improve the patient's sitting posture. Botulinum toxin injections can be employed to treat spasticity. Special education programs and interventions can be considered.[10][11]
Differential Diagnosis
Consider other dysmyelinating diseases such as:
- Adrenoleukodystrophy: typically spares subcortical U-fibers, has a characteristic occipitoparietal periventricular white matter distribution
- Alexander disease: anterior dominance, diffuse late in the disease course, normal NAA on MRS levels
- Metachromatic leukodystrophy: spares subcortical U-fibers, will have a characteristic 'butterfly' pattern
- Pelizaeus-Merzbacher disease: associated with cerebellar atrophy, tigroid pattern of hyperintensity, typically spares subcortical U-fibers
Prognosis
For severe disease, the prognosis is grave. Survival beyond the first decade of life is not always possible. For mild disease, the life expectancy varies. Most patients make it through puberty and may have an average lifespan.
Complications
- Intellectual disability
- Lack of motor skills
- Difficulty feeding
- Hypotonia
- Paralysis
- Blindness
- Seizures
- Death
Pearls and Other Issues
Canavan disease patients have a variable average lifespan. Most people with the neonatal or infantile form survive only into childhood, although some studies report life expectancy up to adolescence or beyond. People with the mild or juvenile form do not appear to have a shortened lifespan.
Enhancing Healthcare Team Outcomes
Canavan disease is a rare lethal neurological disorder which is best managed by an interprofessional team that includes a neurologist, internist, geneticist, social worker, nurse, physical therapist, ophthalmologist, and a gastroenterologist. These patients tend to have many physical limitations including an inability to eat, speak or even walk. The prognosis for most patients is poor, even those with mild disease have a poor quality of life.[12]