Continuing Education Activity
Pityriasis lichenoides encompasses a group of inflammatory cutaneous disorders known as pityriasis lichenoides chronica (PLC), pityriasis lichenoides et varioliformis acuta (PLEVA), and febrile ulceronecrotic Mucha-Habermann disease (a subtype of PLEVA). PLEVA is a relatively benign cutaneous disorder of unclear origin that affects children and young adults. Patients present with a spontaneous eruption of erythematous macules that evolve into papules, rapidly undergoing hemorrhagic necrosis and ulceration. The disease tends to wax and wane with complete resolution, varying in weeks to years. This activity describes the pathophysiology and clinical presentation of PLEVA and the importance of the interprofessional team in recognizing and managing patients with this condition.
Objectives:
- Describe the pathophysiology of pityriasis lichenoides et varioliformis acuta.
- Recall the presentation of pityriasis lichenoides et varioliformis acuta.
- Summarize the treatment and management options for pityriasis lichenoides et varioliformis acuta.
- Review various differential diagnoses for pityriasis lichenoides et varioliformis acuta and associated complications.
Introduction
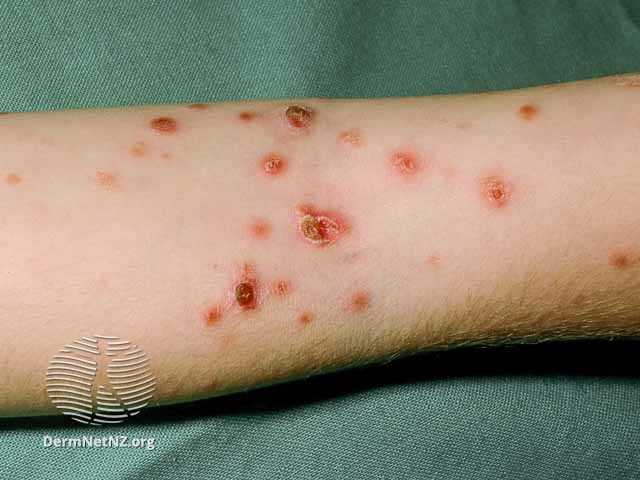
Pityriasis lichenoides et varioliformis acuta (PLEVA), also known as Mucha-Habermann disease, is an uncommon cutaneous inflammatory rash characterized by diffuse red-brown papules in various stages with a mica-like scale on more established lesions. The papules may progress to form vesicles, pustules, and ulcers, and these lesions can be associated with pruritus or a burning sensation. PLEVA favors the trunk and proximal extremities, especially in the flexural regions. This rash tends to relapse and remit with variable duration, sometimes lasting up to years.[1]
Etiology
The exact etiology of PLEVA is not known, although there have been various hypotheses noted in the literature, including an atypical immune response to infections, an inflammatory reaction to medications, and a lymphoproliferative disorder. In support of these hypotheses, there have been cases reported of PLEVA following infections with pathogens such as Epstein-Barr virus, human immunodeficiency virus (HIV), varicella-zoster virus, Toxoplasma gondii, Group A streptococcus, and herpes simplex virus type 2.[1][2]
Additionally, PLEVA has been found to erupt in response to receiving the human papillomavirus, influenza, tetanus, measles, mumps, and rubella (MMR) vaccinations.[3][4][5][6] Due to the risk of development of lymphoma and clinical similarity to lymphomatoid papulosis, the hypothesis regarding lymphoproliferation and T cell dyscrasia must be considered. PLEVA may also develop following the use of certain medications, not limited to atezolizumab, pembrolizumab, and topical diphenylcyclopropenone.[7][8]
Epidemiology
The incidence, prevalence, and risk factors of PLEVA are unknown. The disease does not discriminate by race or gender and mainly affects children and young adults, with a slight male predominance.[1]
Pathophysiology
Due to the ambiguity of the etiology of PLEVA, there are various theories regarding the pathophysiology. Some patients experience deposition of IgM and C3 in the skin lesions and elevated immune complexes in the serum, indicating immune complex or hypersensitivity reaction.[9]
Despite these findings, PLEVA is not considered a vasculitic phenomenon, as fibrin is lacking in the walls of vessels, and thrombi are not present in the lumen. Other lesions exhibit a cell-mediated mechanism with cytotoxic T-cell collections in the dermis, along with decreases in Langerhans cells and the CD4/CD8 ratio.[9]
Additionally, amplifying T cell receptor genes via polymerase chain reaction (PCR) has shown T cell clonality within the lymphocytic infiltrates, suggesting lymphoproliferation with neoplastic potential.[1]
Histopathology
A definitive diagnosis of PLEVA requires a thorough history and physical exam along with a skin biopsy due to many diseases with similar clinical presentations. There are many histopathologic findings for PLEVA, which aid in confirming the diagnosis.[10] Epidermal findings include spongiosis, dyskeratosis, parakeratosis, acanthosis, a few intraepidermal vesicles, and necrosis.[1]
Dermal findings include edema, wedge-shaped superficial and deep dermal lymphohistiocytic inflammatory infiltrate, lymphocyte and erythrocyte extravasation, subepidermal vesicles, and even dermal sclerosis.[9] Vascular findings are notable for papillary dermis blood vessel dilation and engorgement with endothelial proliferation, vascular congestion, and erythrocyte extravasation.[1]
Dermatoscopic findings for PLEVA have been reported in the literature and include a ring of pinpoint vasculature, brown amorphous areas, white scale, and focal melanin deposition within the stratum corneum.[11] Early-phase lesions have crusted brown and amorphous areas, while late-phase lesions contain a central white patch. In both phases, the lesions are encircled by a ring of pinpoint or linear vascular structures.[12]
History and Physical
A thorough history and physical exam can have significant value in the diagnosis of PLEVA due to the nature of the disease and uncertainty regarding its etiology. It is in the provider's best interest to ask patients about recent vaccinations, illnesses, and medications.
On physical exam, PLEVA manifests as an abrupt eruption of 10-50 erythematous macules that quickly form into inflammatory papules with a fine scale. As the rash progresses, the scale tends to remain attached centrally but lifts at the periphery. The papules may then evolve into pityriasis lichenoides chronica (PLC) lesions or undergo hemorrhagic necrosis and become ulcerated, forming the appearance of red-brown crusts, with possible post-inflammatory hyper- and hypopigmentation.[9] In rare cases, darker-skinned patients may experience diffuse macular hypopigmentation instead of the classic papular morphology.[13][14]
PLEVA may be asymptomatic or present with burning and pruritus. The polymorphous lesions tend to favor the trunk and flexural surfaces of the proximal extremities, but a diffuse presentation is not uncommon. PLEVA often occurs in a remitting and relapsing nature that may last several weeks to years before resolution.[15]
Although rare, febrile ulceronecrotic Mucha-Habermann disease (FUMHD) may follow an existing diagnosis of PLEVA or occur de novo. It is important to be aware of this subtype of PLEVA due to the severity and potential lethality of the disease. Patients affected by FUMHD present with necrotic papules that progress to plaques and large coalescent ulcers. Skin ulceration can be extensive and painful, including oral and genital mucosal.[16] Common systemic symptoms include high fever, abdominal pain, diarrhea, central nervous system manifestations, joint pain, respiratory complications, and death.[16]
Evaluation
Although the diagnosis of PLEVA is mainly clinical and aided by dermatoscopy, a histologic evaluation of lesional skin is imperative due to the numerous differential diagnoses with similar presentations. Punch biopsies are recommended to ensure that the epidermis and dermis can be appropriately evaluated. Immunohistochemical studies can aid in confirming the diagnosis and typically show CD8-positive T lymphocytes along with negative CD30 stains (to rule out lymphomatoid papulosis).[9]
Serological studies are not specific for PLEVA, but findings, including leukocytosis, elevated C-reactive protein, and erythrocyte sedimentation rate, can be seen in FUMHD.[1] Other common/useful non-specific tests to diagnose PLEVA by exclusion include toxoplasma Sabin-Feldman dye, heterophile antibody test, antistreptolysin o titers, throat cultures, HIV screening, and rapid plasma reagin test.[1]
Treatment / Management
Due to the benign nature of PLEVA, pharmacologic therapy is unnecessary but may quicken the recovery process and provide relief if the patient is symptomatic. Data regarding treatment for the disease is limited, although various retrospective series and case reports recognize systemic antibiotics and/or phototherapy as first-line therapies in conjunction with corticosteroids.[17]
Systemic antihistamines may also offer relief from pruritus. Furthermore, large ulcerations of PLEVA may benefit from local wound care.[16] Follow-up monitoring is required in aggressive cases of PLEVA with a waxing and waning course.
Antibiotics known for their anti-inflammatory properties are most commonly utilized, such as doxycycline or minocycline, and erythromycin for children.[17][18] Antibiotics are continued until the resolution of lesions for up to three months, and in some cases, longer. If there is no response, phototherapy is considered.
Ultraviolet B light (UVB) is the most common and first-line phototherapy used, with no significant difference between narrow and broadband UVB. Although the long-term effects of phototherapy are unknown, children who received treatment did not experience any early side effects. In adults, therapy is directed by preference and other existing diseases.[17] Psoralen plus ultraviolet A (PUVA) has been reported as effective in multiple case studies.[17] Ultraviolet A1 has been reported in the literature to aid in the complete resolution of lesions, although it is less accessible and more commonly found in academic centers.[19]
Corticosteroids are commonly utilized as adjunctive therapy, and for patients with refractory disease, methotrexate may be an option.[20][21] Additional therapies for refractory cases include acitretin, dapsone, and cyclosporine.[21]
In contrast to the self-limited course of PLEVA, febrile ulceronecrotic Mucha-Habermann disease is a life-threatening disease that presents therapeutic challenges. Many case studies have shown patients with different responses to the same therapeutic approaches, preventing a gold standard treatment from developing.
FUMHD may be treated with TNF-a inhibitors such as infliximab due to elevated TNF-a levels reported in a previous pediatric case and success in an adult case.[22][23][24] Further studies, such as randomized control trials, are needed to optimally assess the efficacy of the therapies mentioned.
Differential Diagnosis
The differential diagnosis for pityriasis lichenoides et varioliformis acuta includes:
- Langerhans cell histiocytosis
- Gianotti-Crost syndrome
- Generalized arthropod bite reaction
- Disseminated herpes simplex virus
- Varicella Zoster virus
- Cutaneous small vessel vasculitis
- Arthropod bite
- Secondary syphilis
- Polymorphous light reaction
- Generalized folliculitis
- Pityriasis lichenoides chronica
- Lymphomatoid papulosis
- Lichen planus
- Hypopigmented mycosis fungoides
- Papular eczematous dermatitis
- Pityriasis rosea
- Guttate psoriasis
- Urticaria pigmentosa
- Exanthematous drug eruption
Prognosis
Although rare, PLEVA may evolve into the potentially lethal variant, febrile ulceronecrotic Mucha-Habermann disease. There have also been reports of PLEVA linked to cutaneous T-cell lymphoma. Otherwise, the prognosis is typically excellent considering the relatively benign clinical course of PLEVA and the rarity of the associated disorders.[22]
Complications
Due to the enigmatic nature of PLEVA, misdiagnosis is a common complication. Lymphomatoid papulosis, for example, is a skin disease that mimics PLEVA, but the separation of the two must be emphasized because of differences in prognosis. Lymphomatoid papulosis has a significant risk of transforming into a cutaneous malignant lymphoma. Pityriasis Lichenoides (PL)-like mycosis fungoides (MF), is a rare form of MF that also appears similarly to PL during the clinical inspection.[25]
Although, the histopathological features of PL-like MF are more representative of typical MF, including but not limited to Pautrier’s microabscess, haloed lymphocytes, and epidermotropism.[25] It is imperative that histology is utilized when evaluating these patients to distinguish LP and PL-like MF from the various subtypes of PL.
Additionally, extensive studies may be warranted if the course of PLEVA has progressed to PLC and is unresponsive to treatment. Although rare, there have been cases reported of CTCL discovered after PLC diagnosis.[26]
In one specific case, a pediatric patient was histologically diagnosed with PLC at eight months and went untreated. He presented to the clinic again at two years of age with the development of an indurated plaque which was confirmed as lymphomatoid papulosis after a second biopsy. The patient again went untreated with the assumption that the rash would spontaneously resolve. With no resolution of the skin disease, the patient was re-evaluated at four years old with an additional biopsy, immunostaining, and polymerase chain reaction analysis for clonal TCR gene rearrangement, which confirmed CTCL involvement.[26]
Concerning outward appearance, the ulceronecrotic lesions in PLEVA may lead to alteration in pigmentation of the skin or heal with varioliform scarring. PLEVA can progress into PLC or present in the more fulminant form, FUMHD. The psychological component of PLEVA must not be overlooked due to its diffuse involvement in exposed skin areas.
Deterrence and Patient Education
In most patients, pityriasis lichenoides et varioliformis acuta is a self-resolving, benign skin disease that typically lasts a few weeks. Patients and their families should be educated that the disease is not contagious, and if symptoms worsen, reassessment is necessary.
Enhancing Healthcare Team Outcomes
An interprofessional team of clinicians, nurses, and pharmacists should work closely with the patient and family members to obtain a thorough history and physical examination for proper diagnosis. Due to the self-resolving nature of pityriasis lichenoides et varioliformis acuta, follow-up visits should occur every three to six months if there is no desire for treatment.
Office visits are useful for reevaluating and ensuring no further disease progression or diagnosis change. Patients who choose to receive treatment, such as antibiotics, phototherapy, or corticosteroids, should be monitored closely to assess efficacy.
Nursing can serve as a liaison contact point for the treating clinicians, specialists, and other healthcare providers, providing patient counseling and keeping all parties informed of changes in patient status. The pharmacist can ensure proper dosing and screen for interactions when pharmaceutical care is opted for, reporting any concerns to the nurse of prescribing clinicians. This interprofessional approach will yield the best patient outcomes. [Level 5]