Continuing Education Activity
Tyrosine is an essential aromatic amino acid required for catecholamines, thyroid hormones, and melanin biosynthesis. Hypertyrosinemia, or increased blood tyrosine level, results from an abnormality in tyrosine metabolism. Various acquired and genetic disorders are known to cause hypertyrosinemia. Congenital deficiency of one of the enzymes involved in the tyrosine catabolism pathway, immaturity of these enzymes in neonates, or hepatocellular dysfunction can lead to hypertyrosinemia. Accumulation of tyrosine and its toxic metabolites are mainly responsible for disease manifestation. This activity reviews the etiopathogenesis, clinical presentation, evaluation, and management of different types of hypertyrosinemia and emphasizes the role of an interprofessional team in the care of affected patients.
Objectives:
Describe different causes of hypertyrosinemia.
Explain etiopathogenesis and modes of presentation of the different types of hypertyrosinemia.
Outline management strategies of hypertyrosinemia.
Summarize genetic counseling and the role of an interprofessional team approach suited for the care of affected individuals.
Introduction
Tyrosine is an essential aromatic amino acid required for catecholamines, thyroid hormones, and melanin biosynthesis. Hypertyrosinemia or increased blood tyrosine levels result from an abnormality in tyrosine metabolism. Various acquired and genetic disorders are known to cause hypertyrosinemia. Congenital deficiency of one of the enzymes involved in tyrosine catabolism, immaturity of these enzymes in neonates, or hepatocellular dysfunction can lead to hypertyrosinemia. Accumulation of tyrosine and its toxic metabolites are mainly responsible for disease manifestation.[1]
Acquired forms of hypertyrosinemia are transient, as in neonates, or secondary to liver disease from a different etiology. Hereditary forms are usually more severe and, if untreated, can lead to liver failure, renal tubular acidosis, hepatocellular carcinoma, coagulopathy, seizures, developmental delay, and neurological crisis.[2]
Normal serum tyrosine level ranges from 30 to 120 micromoles/L. However, symptoms do not manifest until serum levels rise above 500 micromoles/L. In most states in the U.S., hereditary tyrosinemias are detected in the newborn screening (NBS) program, which allows for early diagnosis. However, in developing countries, most cases are diagnosed late. Treatment depends on the etiology. Hereditary types would require lifelong tyrosine-restricted diets. Nitisinone, a drug that inhibits toxic metabolite formation, may also be necessary.[3] Liver transplantation is warranted in cases not responding to medical management.[4]
Etiology
Hypertyrosinemia is broadly divided into two categories:
1. Congenital enzyme deficiency
- Hereditary tyrosinemia type 1 (HT2)
- Hereditary tyrosinemia type 2 (HT2)
- Hereditary tyrosinemia type 3 (HT3)
2. Acquired Hypertyrosinemia
- Transient tyrosinemia of the newborn
- Hypertyrosinemia due to liver disease
- Miscellaneous: Hyperthyroidism, scurvy
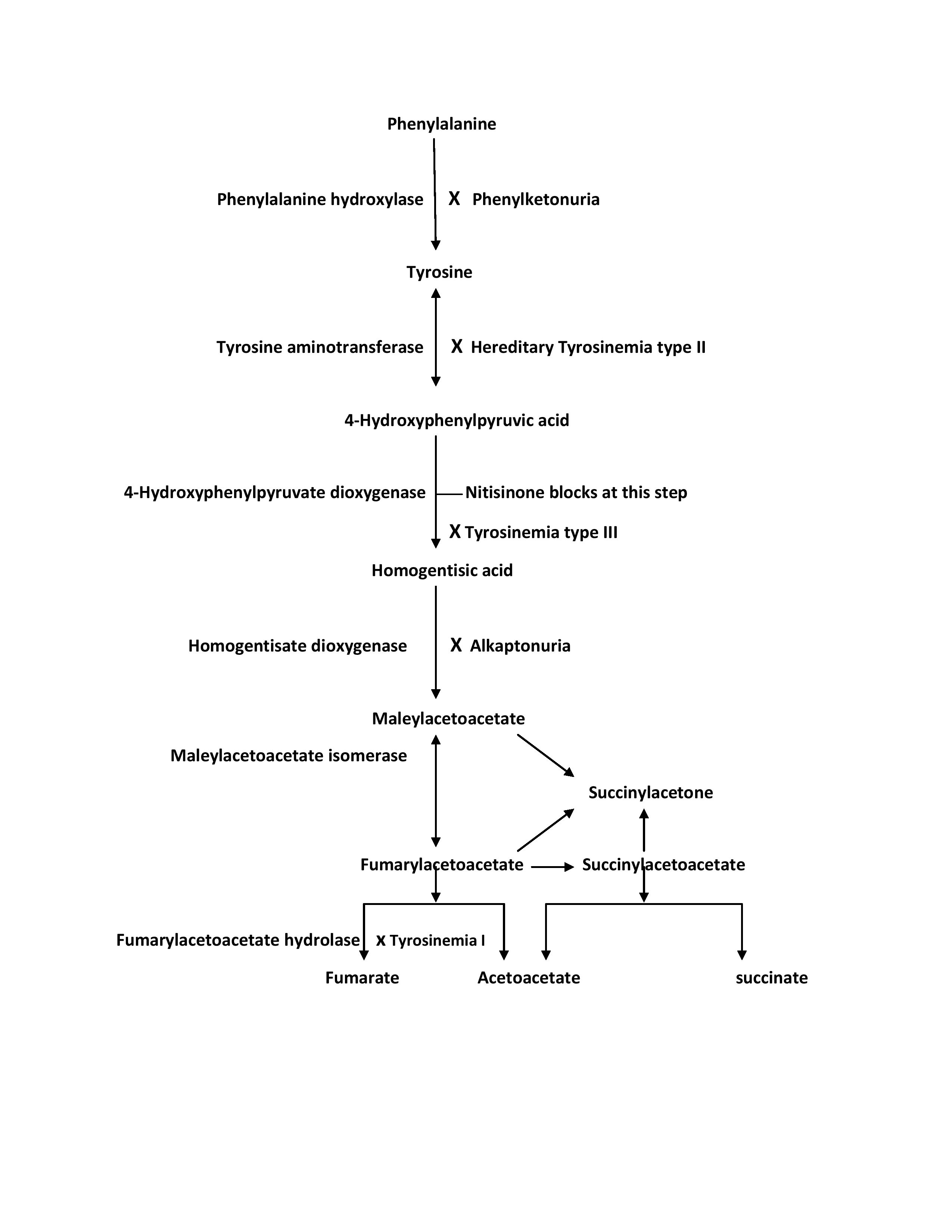
Hereditary tyrosinemia type 1, also known as hepatorenal tyrosinemia, is an autosomal recessive disorder caused by a deficiency of the enzyme fumarylacetoacetate hydrolase (FAH). Mutations in the FAH gene located on locus 15q23-q25 are responsible for this disease.[5] FAH enzyme deficiency leads to fumarylacetoacetate (FAA) accumulation, which gets deposited in hepatocytes and proximal renal tubules, causing liver and kidney damage. FAA is also mutagenic and is believed to cause hepatocellular carcinoma seen in HT1 patients.[6]
The buildup of FAA decreases tyrosine aminotransferase (TAT) activity, the first enzyme in tyrosine metabolism (as shown in the image). This eventually results in elevated tyrosine levels. However, tyrosine levels are not as high as in HT2 patients. FAA has a short half-life and quickly degrades to succinyl acetoacetate (SAA) and succinylacetone (SA). The elevated levels of these metabolites may aid the diagnosis and are utilized in the NBS programs.[7] See Diagram. Tyrosine Metabolism Pathway
Hereditary tyrosinemia type 2 or oculocutaneous tyrosinemia is also known as Richner-Hanhart syndrome. It is an autosomal recessive disorder caused by a deficiency of the enzyme TAT.[8] Mutations in genes encoding TAT on chromosome 16q22 are responsible for this disease.[9] Fifteen different mutations in the TAT gene are known.[10] TAT deficiency results in raised serum levels of tyrosine and its metabolites and is responsible for disease manifestation. The highest serum levels of tyrosine are seen in this type of tyrosinemia. Elevated tyrosine level is utilized for the diagnosis and state newborn screening tests. HT2 patients also demonstrate increased p-hydroxyphenylpyruvate, p-hydroxyphenyllactate, and p-hydroxyphenylacetate in their urine.
Hereditary tyrosinemia type 3 is the rarest of the three types of inherited tyrosinemia, caused by a deficiency of 4-hydroxyphenylpyruvate dioxygenase (HPD).[11] It is also inherited as an autosomal recessive trait and results from a mutation in the HPD gene located on chromosome 12q24.[12] HPD enzyme deficiency leads to an elevation in serum tyrosine levels, which is the marker of this disease. The levels, however, are seldom higher than 500 micromoles/L.
Acquired forms of hypertyrosinemia are notably more common than inherited varieties. Transient tyrosinemia of the newborn is the most common cause of hypertyrosinemia encountered in clinical practice.[13] It is more commonly seen in preterm infants and results from delayed maturation of HPD (the enzyme responsible for HT3) or liver immaturity. The condition is usually asymptomatic and presents with a positive NBS due to an elevated blood tyrosine level. As the name suggests, this entity is transient, and tyrosine levels return to normal within a few weeks. If the levels fail to normalize, work-up for hereditary tyrosinemia is warranted. Lastly, liver dysfunction from any etiology can potentially result in elevated tyrosine levels due to impaired activity of liver enzymes responsible for tyrosine catabolism. Tyrosine levels would be expected to normalize with improvement in liver function.
Epidemiology
HT1 is the most common form of hereditary hypertyrosinemia, with an incidence of 1 in 100,000 individuals worldwide.[14] However, it occurs more frequently in certain regions. In the Saguenay-Lac-Saint-Jean region of Quebec, Canada, the incidence is reported to be 1 in 1846 live births.[15]
It is also more commonly seen in Norway and Finland. HT2 occurs at a frequency of less than 1 in 250,000 worldwide. The incidence of HT3 is largely unknown, and only a few cases are reported in the literature. The recurrence risk in the sibling of an index case is 25% for all types of hereditary hypertyrosinemia based on the Mendelian inheritance pattern.
History and Physical
The clinical presentation of hypertyrosinemia varies depending on the etiology.
HT1 is the most severe type of hypertyrosinemia and presents in two forms. The acute form presents within the first two months of life. The usual presentation is hepatomegaly, jaundice, bleeding, elevated liver enzymes, and a deranged coagulation profile.[16] Affected patients also develop renal Fanconi syndrome and a typical "cabbage odor" urine.[17]
Without treatment, death from progressive liver failure may ensue in the first two years of life. The chronic form presents with a milder phenotype characterized by chronic liver disease and hypophosphatemic rickets related to renal Fanconi syndrome. Other features described are failure to thrive, hypertrophic cardiomyopathy, peripheral neuropathy, and respiratory failure.
HT2 typically presents with eye and skin involvement in the first year of life. Eye symptoms include photophobia and excessive lacrimation due to dendritic keratitis caused by the deposition of tyrosine crystal over the cornea. Skin lesions comprise tender hyperkeratotic plaques involving palms, soles, elbows, knees, and ankles. Almost one-half of the patients with HT2 also have varying degrees of intellectual disability.[18] Unlike HT1, the liver and kidneys are spared in HT2, which helps clinically differentiate this entity from the more severe type 1 hereditary tyrosinemia.
Patients with HT3 present with neurological symptoms ranging from ataxia to seizures and intellectual disability. An HT3 patient with a normal phenotype has also been reported in the literature.[19] Transient tyrosinemia of the newborn is usually asymptomatic and presents only with an abnormal NBS owing to elevated blood tyrosine levels. Similarly, hypertyrosinemia related to liver dysfunction results in mildly elevated tyrosine levels and is usually asymptomatic. However, the presence of liver disease may pose a diagnostic dilemma.[20]
Evaluation
In all 50 states in the U.S. and a few other countries where the incidence of this disease is high, NBS for inherited types of hypertyrosinemia is available. Unfortunately, most other nations do not have a robust NBS program that often allows this disease to go undiagnosed until later. NBS programs rely on tandem mass spectroscopy to detect SA in HT1 or elevated tyrosine levels in HT2 and HT3. Patients suspected of hypertyrosinemia based on clinical features or an abnormal NBS should undergo confirmatory tests for a specific type of tyrosinemia. For example, HT1 patients have elevated SA in their urine or blood and moderate tyrosine elevations. Elevated SA is considered pathognomonic of HT1. These patients also have elevated alpha-fetoprotein (AFP), abnormal liver function, and coagulation profiles.
Patients with HT2 have substantially higher levels of tyrosine (>1000 micromoles/L). In addition, urine from these patients contains large quantities of p-hydroxyphenylpyruvate, p-hydroxyphenyllactate, and p-hydroxyphenylacetate. Patients with HT3 have elevated serum tyrosine levels, but the levels are typically less than 500 micromol/L.
Molecular testing for specific gene mutations is also available for all three types of hereditary hypertyrosinemia. Acquired types of tyrosinemia usually have mild elevations in serum tyrosine levels and typically show up as abnormal NBS in the case of transient tyrosinemia of the newborn. Since the tyrosine levels are only mildly elevated, these patients are clinically asymptomatic.
Once a diagnosis has been made, most patients with HT1 and HT2 require interdisciplinary team management involving a pediatrician/neonatologist and a gastroenterologist, cardiologist, neurologist, ophthalmologist, dermatologist, a medical geneticist from the pediatric specialty along with a nutritionist experienced in managing hypertyrosinemia patients. Timely consultations and regular follow-ups with these sub-specialties cannot be overemphasized. Affected HT1 patients would also need abdominal imaging to rule out hepatocellular carcinoma. HT2 patients would benefit from echocardiography to rule out cardiomyopathy and nerve conduction studies to evaluate peripheral neuropathy.[21]
In terms of imaging, all patients with features to suggest hypertyrosinemia should get an ultrasound of the liver and kidneys. If there are nodules in the liver, further imaging, preferably an MRI, should be carried out. A bone radiograph of the wrist or chest should be done in cases of confirmed tubulopathy.
Treatment / Management
Treatment of hypertyrosinemia depends upon the etiology and aims to limit tyrosine intake and block the formation of toxic tyrosine metabolites. Since untreated HT1 can have a fulminant course, delaying treatment until a confirmatory test is not advised, a diet low in protein, along with nitisinone, should be started as soon as possible. Nitisinone or NTBC (2-nitro-4-trifluoromethylbenzoyl 1,3-cyclohexanedione) is an FDA-approved drug for the treatment of HT1. It inhibits the HPD enzyme, thereby limiting the formation of toxic tyrosine metabolites.[22] The clinical efficacy of nitisinone has been demonstrated in multiple clinical trials.[23][24] When started early in the course, it has been shown to reduce the risk of hepatocellular carcinoma.[24]
Nitisinone increases blood tyrosine levels in patients with HT1 by blocking its catabolism. Excess tyrosine may deposit over the corneas causing corneal ulcerations. Therefore, it is important to restrict the dietary intake of tyrosine while patients are on nitisinone. Some patients on prolonged nitisinone therapy combined with dietary protein restriction may develop phenylalanine deficiency. This deficiency has been linked to neurodevelopmental issues in HT1 patients.[25]
A recent study has reported positive long-term therapeutic and safety assessments.[26] Nitisinone should be used in conjunction with dietary modification regarding tyrosine and phenylalanine. Management with nitisinone and dietary modification should begin as early as possible after the confirmation of the diagnosis. There is recent evidence to suggest that tyrosine tolerance increases slightly with age, although this should not mean that there is no need for close monitoring.[27]
Therefore, phenylalanine supplementation is recommended during nitisinone therapy. Few patients on nitisinone treatment may develop transient neutropenia and thrombocytopenia, the only known adverse drug reaction of this drug. Long-term monitoring of HT1 patients is done by serial measurement of plasma amino acids, SA levels, liver and kidney functions, serum AFP levels, complete blood count, and annual abdominal imaging. The small subset of patients who remain refractory to medical treatment and show signs of persistent liver failure or hepatocellular carcinoma would benefit from orthotopic liver transplantation.
Treatment of HT2 involves keeping serum tyrosine levels below 500 micromoles/L and is accomplished by dietary tyrosine and phenylalanine restriction. The skin and eye lesions respond dramatically and normalize completely within weeks of this therapy.[18] HT2 patients may also benefit from vitamin C supplementation. Similarly, for patients diagnosed with HT3, dietary restriction of tyrosine and phenylalanine is recommended. However, it is not known if this therapy would prevent or reverse neurological abnormalities seen in HT3 patients. Low tyrosine and phenylalanine formulas are commercially available for use in hypertyrosinemia patients.
Differential Diagnosis
Clinical presentation of HT1 may mimic neonatal sepsis, liver failure from any other etiology, cystinosis, Lowe syndrome, or hereditary fructose intolerance.[28] A high index of suspicion, careful history, physical exam, and appropriate laboratory test will help delineate the diagnosis in doubtful cases. Patients with neonatal sepsis have abnormal CBC, C-reactive protein, or positive cultures and respond to antibiotics. Cystinosis is an autosomal recessive lysosomal disorder with abnormal transport of amino acid cysteine.
The nephropathic form of cystinosis typically presents within the infantile period with features of Fanconi syndrome, hypophosphatemic rickets, hyperpigmentation, and deposits over the cornea seen on slit-lamp examination. Diagnosis of cystinosis is usually made by demonstrating elevated cysteine levels in leukocytes or fibroblasts. Lowe syndrome, also known as the oculocerebrorenal syndrome, is an X-linked recessive disorder and presents during the first year of life with bilateral congenital cataracts, hypotonia, and renal Fanconi syndrome in male probands.[29] Clinically it is differentiated from HT1 by the presence of cataracts, and hypotonia noted at birth. In doubtful cases, diagnosis can be confirmed by molecular genetic testing.
Lastly, hereditary fructose intolerance can also present with renal Fanconi syndrome. Still, it can easily be differentiated from HT1 by the temporal association of symptoms with the introduction of fructose or sucrose in the diet. Diagnosis can be confirmed by sequencing the Aldolase B gene.[30]
HT2 patients present with corneal ulcerations and dermal keratosis and can be confused with other causes of corneal ulcerations in infancy, especially in regions where NBS for this disease is unavailable. Infectious causes (both bacterial and viral) can cause corneal ulcers in infants. Patients with infectious causes usually have other signs and symptoms of sepsis and will respond to appropriate antimicrobial agents. Differential diagnosis of HT3 includes causes of ataxia, seizures, and psychomotor retardation. A careful history, physical exam, and detection of elevated tyrosine levels will aid in the diagnosis of HT3 in doubtful cases.
Prognosis
The prognosis depends on the etiology. In HT1, where treatment is started early, the prognosis tends to be better. One study in which five HT1 patients were started on nitisinone and dietary restriction within 28 days of life resulted in no death or liver failure during 15 years of follow-up.[31]
Untreated patients may succumb to acute liver failure before two years of life or from chronic liver failure and hepatocellular carcinoma during the second decade of life. HT2 and HT3 are rare diseases, and long-term survival data are unknown. However, both of these diseases usually have a milder course than HT1. Furthermore, the outcome is significantly improved by early diagnosis and treatment. The introduction of the NBS program in many developed countries has been instrumental in reducing the morbidity and mortality associated with these disorders.
Complications
The complications of untreated or poorly treated HT1 are liver failure, cirrhosis, and hepatocellular carcinoma, resulting in early death. Neurological complications are also common in HT1 patients, ranging from porphyria-like neuropathic crisis to muscle weakness, often leading to respiratory failure. Renal Fanconi syndrome can result in hypophosphatemic rickets.
HT2 is typically milder than HT1, and complications in untreated patients are related to corneal ulcerations and painful keratotic plaques. However, 50% of HT2 patients may also develop some degree of intellectual disability.[18] Untreated HT3 patients can have debilitating ataxia and intractable seizures. Intellectual disability is also common in HT3.
Consultations
Patients suspected or diagnosed with any type of hereditary hypertyrosinemia would need an interprofessional team approach for optimizing management. Primary care physicians will play a central role in collaborating care with the following sub-specialists:
- Medical geneticist
- Pediatric gastroenterologist
- Pediatric nephrologist
- Pediatric cardiologist
- Pediatric neurologist
- Pediatric ophthalmologists and pediatric dermatologists in patients with HT2
- Nutritionist with experience in managing hypertyrosinemia
Deterrence and Patient Education
Detailed counseling, including advice on dietary restrictions and treatment adherence, is vital to improving the long-term outcome. In addition, patients and families need to be educated on the complications of the disease and the signs to monitor. Information about possible side effects of therapy should be given as well. Families would also benefit from appropriate genetic counseling for pregnancy planning.
Enhancing Healthcare Team Outcomes
Most cases of hereditary tyrosinemia in the western world are diagnosed with newborn screening. However, newborn screening remains a distant reality in developing countries, and most patients receive a late diagnosis. It has been shown that early diagnosis and treatment improve the long-term outcome of affected individuals.
These disorders involve multiple systems, and optimal management requires an interprofessional team approach involving clinicians (MDs, DOs, NPS, and PAs), nurses, lab technicians, pharmacists, nutritionists, and physicians from various sub-specialties. Clinicians will guide care, but nurses will play a pivotal role in patient care by assisting in patient evaluation, patient counseling and serving as a liaison between the various providers on the case. Pharmacists and dieticians or nutritionists can round out the team and must communicate their concerns or findings with other team members promptly. All care team members must maintain accurate and updated patient records. This interprofessional approach will yield optimal patient results. [Level 5]
Every healthcare professional involved in the care of these patients needs to be aware of the current evidence-based management approaches, especially given the condition's rarity. Adherence to dietary restriction and nitisinone therapy (for HT1) remains the mainstay of treatment and needs to be instituted early for the best outcome. [Level 3]