Continuing Education Activity
Biosimilars is FDA approved for the treatment of rheumatoid arthritis (RA), polyarticular juvenile idiopathic arthritis (JIA), psoriatic arthritis (PA), ankylosing spondylitis (AS), plaque psoriasis (PsO), inflammatory bowel disease (IBD): adult Crohn disease (CD), ulcerative colitis (UC), granulomatosis with polyangiitis (GPA) (Wegener granulomatosis), microscopic polyangiitis (MPA), type 1 diabetes mellitus (DM), non-squamous non-small cell lung cancer, metastatic colorectal cancer, metastatic renal cell carcinoma, glioblastoma, recurrent or metastatic cervical cancer, HER2 (human epidermal growth factor receptor 2) associated breast cancer, and HER2 associated gastric (metastatic) or gastroesophageal junction adenocarcinoma, chronic lymphocytic leukemia (CLL), non-Hodgkin lymphoma (NHL). Biosimilars are biotherapeutic agents similar to their respective original biologic product (reference product). This activity will highlight the mechanism of action, adverse event profile, and other key factors pertinent to interprofessional team members in managing patients with inflammatory diseases and the use of biosimilars.
Objectives:
- Identify the mechanism of action of all biosimilars.
- Identify the indications for the use of biosimilars.
- Identify the issues of concern that exist for biosimilars.
- Summarize interprofessional team strategies for improving care coordination and communication to advance biosimilars use in treating patients with inflammatory diseases and improve outcomes.
Introduction
Biosimilars are biological agents that are highly analogous to their reference products currently approved and licensed by the U.S. Food and Drug Administration (FDA). Biosimilars possess no differences in their safety, purity, potency, or effectiveness compared to their respective reference biologic agents. The abbreviated licensure pathway 351(k) for biosimilars or agents interchangeable with FDA-approved biological agents (reference product) was formed by Congress by The Biologics Price Competition and Innovation Act (BPCI Act) of 2009.[1] Biosimilars aim to increase the number of biologics available at more inexpensive costs when compared to their reference products, further increasing patient access to more treatment options.[2]
Similar to biologics or reference products, biosimilars are expected to meet the FDA's meticulous approval criteria, ensuring the reliability of the agent's safety, efficacy, quality, and effectiveness.[3] The FDA approval process for biosimilars is based on data proving its prominent similarity and correlation to an already FDA-approved biologic agent (reference product) and its indications for use. No clinically meaningful differences among the biosimilar and its respective reference agent should exist.[3] Differences exist in the regulatory provisions for the original biologic agent (reference product) and a biosimilar as the biosimilars obtain approval from FDA by their abbreviated approval pathway.[4] Biosimilars seeking FDA approval must include data assessments demonstrating the agent's similarity to a reference product acquired from analytical investigations, animal studies, and clinical studies. Added data and analyses of the biosimilars immunogenicity, pharmacokinetics, pharmacodynamics, and data discussing the indication(s) the reference product is licensed and approved for use are also included.[5]
To date, ten biological agents: bevacizumab, etanercept, epoetin-alfa, trastuzumab, adalimumab, pegfilgrastim, filgrastim, infliximab, rituximab, and insulin glargine, have been used as reference products for the approval of 30 biosimilar agents. Biosimilars can be used in the management of various inflammatory and autoimmune diseases. Ailments such as rheumatoid arthritis (RA), polyarticular juvenile idiopathic arthritis (JIA), psoriatic arthritis (PA), ankylosing spondylitis (AS), plaque psoriasis (PsO), inflammatory bowel disease (IBD): adult Crohn disease (CD), ulcerative colitis (UC), granulomatosis with polyangiitis (GPA) (Wegener granulomatosis), microscopic polyangiitis (MPA), and type 1 diabetes mellitus (DM). Filgrastim-sndz, a biosimilar of the reference product "filgrastim," was the first-ever biosimilar to receive FDA approval in March 2015 and is characterized as a leukocyte growth factor indicated to reduce infections caused by febrile neutropenia in the neutropenic and immunosuppressed subjects.
Other indications of biosimilars alongside inflammatory diseases include management for specific malignancies and secondary causes of anemia. In September 2017, bevacizumab-awwb was the first biosimilar that was granted FDA approval for certain cancers. A few months later, in December 2017, biosimilar trastuzumab-dkst was also granted FDA approval for specific malignancies. Cancers indicative of biosimilar therapy include non-squamous non-small cell lung cancer, metastatic colorectal cancer, metastatic renal cell carcinoma, glioblastoma, recurrent or metastatic cervical cancer, HER2 (human epidermal growth factor receptor 2) associated breast cancer, and HER2 associated gastric (metastatic) or gastroesophageal junction adenocarcinoma, chronic lymphocytic leukemia (CLL), non-Hodgkin lymphoma (NHL). To date, new biosimilars are currently still being FDA approved, with the latest approval coming on July 28, 2021, for biosimilar agent insulin glargine-yfgn, a long-acting insulin agent indicated for pediatric type 1 diabetes mellitus (DM) and adult patients with DM type 1 and DM type 2.
Biosimilars agent insulin glargine-yfgn is the first insulin biosimilar agent to receive FDA approval and is also the first interchangeable agent in the class of biosimilar agents. Interchangeable products are biosimilars that meet additional criteria specified by the Biologics Price Competition and Innovation Act during the assessment and testing of the agent during its FDA approval process.[6] Data is required demonstrating clinical results as its respective reference product in any subject treated with the interchangeable products.[6] Data on the interchangeable products substituted with their reference product in terms of safety and efficacy of alternating back and forth have been assessed and evaluated by the FDA prior to their approval. Once an interchangeable product has been approved, it may be interchanged for their reference product without requiring additional consulting from the prescribing clinician or healthcare professional.[7][8]
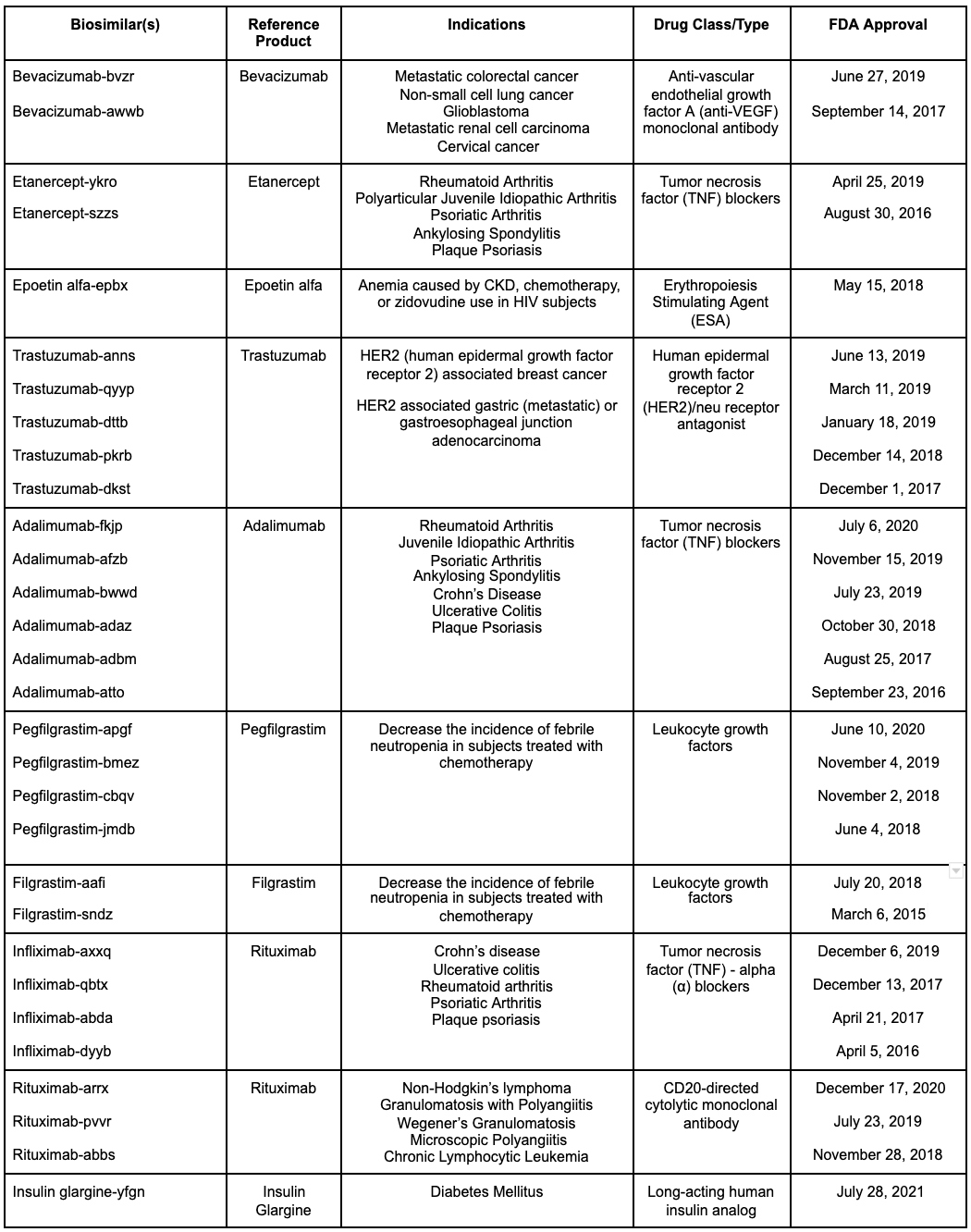
Table 1: Summarizes all FDA approved biosimilars
Function
The biosimilar's mechanism of action mimics and produces effects highly analogous to their reference products and do not pose any clinically significant differentiation concerning safety and effectiveness.[6] Scientific investigations of biosimilars show slight variations in clinically inactive components as they are reversed engineered; their formation is profoundly dependent on the primary component of their respective reference products and shows safety, purity, and potency when treating ailments its reference product is approved and indicated for use.[2] Biosimilars are highly similar but not identical to conventional biologics.
Bevacizumab
Bevacizumab-bvzr and bevacizumab-awwb are classified as anti-vascular endothelial growth factor A (anti-VEGF) monoclonal antibody rendering them biosimilar to their reference product bevacizumab, also a vascular endothelial growth factor inhibitor. Similar to bevacizumab, the biosimilars possess no differences in exerting their mechanism of action, binding VEGF, and hindering its interplay with its target receptors Flt-1 and KDR located on the surface of endothelial cells. The activity further inhibits VEGF-mediated endothelial cell proliferation and preventing new blood vessel formation (angiogenesis).
Although the biosimilar agents are indicated for non-small cell lung cancer, colorectal cancer, glioblastoma multiforme, renal cell carcinoma, and cervical cancer similarly to their reference product (bevacizumab), their indications are not currently approved for the treatment of hepatocellular carcinoma (HCC) and epithelial ovarian, fallopian tube, or primary peritoneal cancer like their reference product (bevacizumab).[9]
Etanercept
Etanercept-ykro and etanercept-szzs are classified as tumor necrosis factor (TNF) blockers making them biosimilar to their reference product etanercept, also a TNF blocker. Alike their reference product (etanercept), biosimilars etanercept-ykro and etanercept-szzs are dimeric soluble formations of the p75 TNF receptor, which exerts their actions by preventing the binding of TNF-alpha (α) and TNF-beta (β), also known as lymphotoxin alpha (LT-α) to their target surface receptors.
The blocking of TNF-α and TNF-β to TNF receptors puts TNF in a biologically inactive state, further decreasing its role in inflammatory processes and immune responses. Increases in TNF are seen in inflammatory diseases such as rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, and plaque psoriasis, making etanercept and its biosimilar products indications use.[10]
Epoetin alfa
Epoetin alfa-epbx is classified as an erythropoiesis-stimulating agent (ESA), rendering them biosimilar to its reference product epoetin alfa, also an ESA. Epoetin alfa-epbx exerts the same mechanism of action as its reference product (epoetin alfa) and endogenous hormone erythropoietin (EPO). The human recombinant EPO, epoetin alfa-epbx, and its reference product (epoetin alfa) comprise the same amino acid sequence as isolated endogenous EPO. The further agents bind the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway binding receptor on the surface of their target cells.
The receptor-binding further signals intracellular phosphorylation activating transcription factors to regulate gene expression, influencing erythropoiesis in a dose-dependent fashion. Epoetin alfa-epbx, and its reference product (epoetin alfa) share identical indications for use in anemia precipitated by chronic kidney disease and chemotherapy and HIV-infected subjects treated with zidovudine causing anemia.[11]
Trastuzumab
Trastuzumab-anns, trastuzumab-qyyp, trastuzumab-dttb, trastuzumab-pkrb, and trastuzumab-dkst are classified as human epidermal growth factor receptor 2 (HER2)/neu receptor antagonist making them biosimilar to their reference product trastuzumab, also a HER2/neu receptor antagonist. Analogous to its reference product (trastuzumab), the biosimilars are a monoclonal antibody directed against HER2 proto-oncogene extracellular receptor and hinder the HER2 homodimerization, further halting its HER2-mediated signaling.
Human tumors cells demonstrating overexpression of HER2, trastuzumab, and its corresponding biosimilars agents have been shown to inhibit proliferation and tumor cell growth and promote antibody-dependent cellular cytotoxicity (ADCC), inducing cell death of HER2 expressing human cells. Trastuzumab biosimilars share the same indications as their reference products, making them useful in treating HER2-overexpressing breast cancer and HER2-overexpressing stomach cancer.[12]
Adalimumab
Adalimumab-fkjp, adalimumab-afzb, adalimumab-bwwd, adalimumab-adaz, adalimumab-adbm, and adalimumab-atto are classified as a tumor necrosis factor (TNF) blocker rendering them biosimilar to their reference product adalimumab, which is also a TNF blocker. Parallel to their reference product (adalimumab), the biosimilars are an immunoglobulin G (IgG) anti-TNF alpha monoclonal antibody that binds respectively to the TNF-alpha, further hindering its interplay with its target cell surface TNF receptors TNFR1(p55) and TNFR2 (p75).
Adalimumab and its biosimilars, although a TNF-inhibitor, the agents do not bind or inactivate TNF-beta (lymphotoxin), as TNF-inhibitor etanercept and its corresponding biosimilars. The TNF inhibition restrains the receptor activator of NF-κB (RANK) ligand (RANKL) protein, further halting the consequent destruction of cartilage and bone, providing its effectiveness in inflammatory arthritic diseases. TNF is an endogenous cytokine present in common inflammatory and immune responses. Increased amounts of TNF are observed in the synovial fluid of subjects with rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, and ankylosing spondylitis, making adalimumab and its corresponding biosimilars indications for its use.
Although the biosimilars of adalimumab exert the same mechanism of action, they differ in their indications for uveitis (UV), hidradenitis suppurativa (HS), and pediatric Crohn disease (CD). As the conventional biologic (adalimumab) is indicated and approved for these uses, the biosimilars are not approved for UV, HS, and pediatrics CD.[13]
Pegfilgrastim
Pegfilgrastim-apgf, pegfilgrastim-bmez, pegfilgrastim-cbqv, and pegfilgrastim-jmdb are classified as leukocyte growth factors making them biosimilar to their reference product pegfilgrastim, also a leukocyte growth factor. Reference agent pegfilgrastim and its comparable biosimilars are granulocyte colony-stimulating factors (G-CSF) exerting their effects by binding distinct target cell surface receptors on hematopoietic cells.
The biosimilars and their reference agent (pegfilgrastim) also halt cell activation and further promote mature neutrophils' survival and are indicated to decrease the occurrence of febrile neutropenia in subjects with cancer receiving chemotherapy. Unlike their reference product (pegfilgrastim), its biosimilars are not indicated and approved for its use in subjects acutely exposed to myelosuppressive dosages of radiation, also known as "hematopoietic subsyndrome of acute radiation syndrome (ARS)."[14]
Filgrastim
Filgrastim-aafi and filgrastim-sndz are classified as leukocyte growth factors rendering them biosimilar to their reference product filgrastim, also a leukocyte growth factor. Like its reference product, filgrastim biosimilars are human recombinant granulocyte colony-stimulating factor (G-CSF) glycoproteins exerting their function by binding to their target receptors located on the cell surface, promoting cell maturation and proliferation of neutrophil progenitors.
Within the body, endogenous G-CS is generated by specific cells such as monocytes‚ fibroblasts, and endothelial cells and functions as a lineage-specific colony-stimulating factor. The G-CSF function inside the bone marrow targets neutrophil progenitor proliferation and coordinates the generation and differentiation of neutrophils and functional end-cell activation and further promotes mature neutrophils' survival, and are indicated to decrease the occurrence of febrile neutropenia in subjects with cancer receiving chemotherapeutics.[15]
Infliximab
Infliximab-axxq, infliximab-qbtx, infliximab-abda, and infliximab-dyyb are classified as tumor necrosis factor (TNF) - alpha (α) blockers making them biosimilar to their reference product infliximab, also a TNF-α blocker. Similar to their reference product (infliximab), the biosimilars are an immunoglobulin G (IgG) anti-TNF-α monoclonal antibody that binds respectively to the soluble and transmembrane forms of TNF-α, further hindering its interplay with its target cell surface TNF receptors TNFR1(p55) and TNFR2 (p75).
Infliximab and its biosimilars, although a TNF inhibitor, the agents do not bind or inactivate TNF-beta (LT-α). The binding and neutralization of TNF-α reduces its biologic function such as initiation of pro-inflammatory cytokines and acute-phase reactants such as interleukin (IL)-1 and IL-6, decreases endothelial layer permeability, further diminishing leukocyte migration, decreases functional action of neutrophils and eosinophils, and decreases synoviocytes and chondrocytes enzymes affecting tissue degradation. Infliximab and its biosimilars exert the same mechanism of action and share the same disease indications such as Crohn disease, pediatric Crohn disease, ulcerative colitis, rheumatoid arthritis, psoriatic arthritis, and plaque psoriasis.[16][17]
Rituximab
Rituximab-arrx, rituximab-pvvr, and rituximab-abbs are classified as CD20-directed cytolytic monoclonal antibodies, rendering them biosimilar to reference product rituximab, also a monoclonal antibody targeting CD20. Rituximab and its biosimilars exert their effects by targeting pre-B-lymphocytes and mature B-lymphocytes expressing the CD20 antigen on its cell surface, further prompting cell lysis of the targeted B-lymphocytes. Cell death/lysis mechanisms include antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-mediated-cytotoxicity (CDC).
Other causes of cell lysis include direct influences of rituximab and its biosimilars when bound to CD20 and antibody-dependent phagocytosis (ADP). B lymphocytes are understood to function in the pathogenesis of rheumatoid arthritis and autoimmune diseases in various methods such as proinflammatory cytokine generation, rheumatoid factor (RF) composition, and decreasing activation of T-lymphocytes and plasma cell production.
Rituximab and its biosimilar's effects on CD20 B cells make them useful and indicated for the treatment of granulomatosis with polyangiitis (GPA) (Wegener granulomatosis), microscopic polyangiitis (MPA), chronic lymphocytic leukemia (CLL), and CD20-positive B-cell non-Hodgkin lymphoma (NHL). Unlike their reference product, the biosimilars are not approved or indicated to treat pemphigus vulgaris (PV).[18]
Insulin glargine
Insulin glargine-yfgn is classified as a long-acting human insulin analog, making it biosimilar to its reference product insulin glargine, also a long-acting human insulin analog.[19] Like its reference product, insulin glargine-yfgn regulates glucose metabolism by binding to its target tyrosine kinase insulin receptors, further causing a conformational transition to the receptors' beta catalytic domains resulting in auto-phosphorylation activation of the beta subunits.[19]
The beta subunits further stimulate phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3k), initiating kinase B, which controls the action of the glucose transporter type 4 (GLUT4) receptor, protein kinase C (PKC), and mitogen-activated protein kinase (MAPK).[19] The intracellular pathway activation further reduces plasma concentration by inciting peripheral glucose uptake by cells in the skeletal muscle and adipose tissue and diminishing glucose production in the liver. It also additionally represses lipolysis and proteolysis and increases protein synthesis. Insulin glargine-yfgn, similar to its reference agent, is indicated in the treatment of type 1 diabetes mellitus.[19]
Issues of Concern
Immunogenicity remains an issue of interest when discussing biotherapeutics. Biosimilars may evoke an immune response (immunogenicity) in subjects receiving therapy like their reference products or original biologics.[20] Immunogenicity to biosimilars can further influence its efficacy and safety and further hamper the evaluation of their comparability to biologics and delay the development and progression of biosimilars.[21]
Assessing the immunogenicity of biosimilars during their clinical development phases and post-marketing monitoring is essential.[5] Immunogenicity is defined as the development of anti-drug antibodies (ADAs) after receiving treatment with a biologic or biosimilar, which may further result in hypersensitivity reactions and/or diminished drug efficacy.[21]
Anti-drug antibodies binding to the biologic or biosimilar active site and further repressing their action are known as neutralizing antibodies.[8] Non-neutralizing antibodies that do not bind the biotherapeutics active site may still result in clinical manifestations, such as reducing drug bioavailability further diminishing biologic or biosimilar therapeutic efficacy.[8]
Neutralizing or non-neutralizing anti-drug antibodies formation is frequently understood as the underlying mechanism demonstrating the decreased efficacy or treatment failure of some biologics.[8] Although the presence of neutralizing or non-neutralizing anti-drug antibodies does not explicitly render clinically meaningful consequences in terms of safety or efficacy, undesired immunogenicity can establish a serious barrier in the development of future biosimilars.[8]
Clinical Significance
To date, the FDA has approved thirty biosimilar agents from 10 reference products:
Reference drug: Bevacizumab
Biosimilar(s)[9]
- Bevacizumab-bvzr (FDA-approved in June 2019)
- Bevacizumab-awwb (FDA-approved in September 2017)
Indications for biosimilar:
- Metastatic colorectal cancer, with intravenous 5-fluorouracil–based chemotherapy (first-line or second-line therapy).
- Metastatic colorectal cancer, with fluoropyrimidine-irinotecan- or fluoropyrimidine-oxaliplatin-based chemotherapy (second-line therapy in subjects who have progressed on a first-line bevacizumab product-containing regimen).
- Non-squamous non-small cell lung cancer (non-resectable, locally advanced, recurrent, or metastatic disease) in sequence with carboplatin and paclitaxel (first-line therapy).
- Glioblastoma, as monotherapy for adult subjects with progressive disease course following prior therapy.
- Metastatic renal cell carcinoma in combination therapy with interferon alfa.
- Cervical cancer (persistent, recurrent, or metastatic), in sequence with paclitaxel and cisplatin or paclitaxel and topotecan.
Reference drug: Etanercept
Biosimilar(s)[10]
- Etanercept-ykro (FDA-approved in April 2019)
- Etanercept-szzs (FDA-approved in August 2016)
Indications for biosimilar:
- Rheumatoid arthritis
- Polyarticular juvenile idiopathic arthritis (subjects 2 years and older)
- Psoriatic arthritis
- Ankylosing spondylitis
- Plaque psoriasis (subjects 4 years and older)
Reference drug: Epoetin-alfa
Biosimilar(s)[11]
- epoetin alfa-epbx (FDA-approved in May 2018)
Indications of Biosimilar:
- Anemia due to chronic kidney disease (CKD) regardless of dialysis status.
- Anemia from zidovudine use in HIV-infected subjects.
- Anemia due to concurrent use of myelosuppressive chemotherapy (once initiated, at least eight weeks of planned chemotherapy).
- Anemia from the decline of allogeneic RBC transfusions in subjects enduring elective, noncardiac, nonvascular surgery.
Reference drug: Trastuzumab
Biosimilar(s)[12]
- Trastuzumab-anns (FDA-approved in June 2019)
- Trastuzumab-qyyp (FDA-approved in March 2019)
- Trastuzumab-dttb (FDA-approved in January 2019)
- Trastuzumab-pkrb (FDA-approved in December 2018)
- Trastuzumab-dkst (FDA-approved in December 2017)
Indications of Biosimilar:
- HER2 (human epidermal growth factor receptor 2) associated breast cancer
- HER2 associated gastric (metastatic) or gastroesophageal junction adenocarcinoma
Reference drug: Adalimumab
Biosimilar(s)[13]
- Adalimumab-fkjp (FDA-approved in July 2020)
- Adalimumab-afzb (FDA-approved in November 2019)
- Adalimumab-bwwd (FDA-approved in July 2019)
- Adalimumab-adaz (FDA-approved in October 2018)
- Adalimumab-adbm (FDA-approved in August 2017)
- Adalimumab-atto (FDA-approved in September 2016)
Indications for biosimilar:
- Rheumatoid arthritis
- Juvenile idiopathic arthritis
- Psoriatic arthritis
- Ankylosing spondylitis
- Adult Crohn disease
- Ulcerative colitis
- Plaque psoriasis
Reference drug: Pegfilgrastim
Biosimilar(s)[14]
- Pegfilgrastim-apgf (FDA-approved in June 2020)
- Pegfilgrastim-bmez (FDA-approved in November 2019)
- Pegfilgrastim-cbqv (FDA-approved in November 2018)
- Pegfilgrastim-jmdb (FDA-approved in June 2018)
Indications for biosimilar:
- To reduce the occurrence of infection caused by febrile neutropenia in subjects with non-myeloid cancer receiving myelosuppressive antineoplastic agents that have correlations with a clinically significant occurrence of febrile neutropenia.
Reference drug: Filgrastim
Biosimilar(s)[15]
- Filgrastim-aafi (FDA-approved in July 2018)
- Filgrastim-sndz (FDA-approved in March 2015)
Indications of biosimilar:
- Reduce the occurrence of infection caused by febrile neutropenia in subjects with non-myeloid cancer receiving myelosuppressive antineoplastic agents that are correlated with a clinically significant occurrence of febrile neutropenia.
- Decreasing the duration of neutrophil recovery and the span of fever post-chemotherapy in subjects with acute myeloid leukemia (AML) post-chemotherapy.
- Decreasing the time of neutropenia and neutropenia-associated clinical abnormalities in subjects with non-myeloid cancers receiving myeloablative chemotherapy accompanied by bone marrow transplantation (BMT).
- Decrease the number and extent of severe neutropenia associated abnormalities in symptomatic patients with congenital‚ cyclic, and idiopathic neutropenia.
- Mobilize hematopoietic progenitor cells obtained from the same individual toward the peripheral blood for acquisition by leukapheresis.
Reference drug: Infliximab
Biosimilar(s)[16][17]
- infliximab-axxq (FDA-approved in December 2019)
- infliximab-qbtx (FDA-approved in December 2017)
- infliximab-abda (FDA-approved in May 2017)
- infliximab-dyyb (FDA-approved in April 2016)
Indications for biosimilar:
- Crohn disease
- Pediatric Crohn disease
- Ulcerative colitis
- Rheumatoid arthritis in conjunction with methotrexate
- Psoriatic arthritis
- Plaque psoriasis
Reference drug: Rituximab
Biosimilar(s)[18]
- Rituximab-arrx (FDA-approved in December 2020)
- Rituximab-pvvr (FDA-approved in July 2019)
- Rituximab-abbs (FDA-approved in November 2018)
Indications for biosimilar:
- Granulomatosis with polyangiitis (Wegener Granulomatosis) in adult subjects in conjunction with glucocorticoids.
- Microscopic polyangiitis in adult subjects in conjunction with glucocorticoids.
- Chronic lymphocytic leukemia in adult subjects that were untreated or treated for CD20-positive CLL in sequence with fludarabine and cyclophosphamide (FC).
- Used as solo therapy for low-grade or follicular CD20-positive B-cell non-Hodgkin lymphoma in adult subjects that have relapsed or refractory to treatment.
- Conjunction with first-line chemotherapy for adult patients with follicular, CD20-positive, B-cell non-Hodgkin lymphoma that did not receive therapy earlier.
- Single-agent maintenance therapy for adult patients with follicular, CD20-positive, B-cell non-Hodgkin lymphoma who attained a full or partial response to a rituximab agent in sequence with a chemotherapeutic drug.
- Monotherapy following first-line agents: cyclophosphamide, vincristine, and prednisone (CVP) chemotherapy regimens, for low-grade, CD20-positive, B-cell non-Hodgkin lymphoma that is stable and is not progressing.
- In sequence with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), or other anthracycline-based chemotherapeutic regimens in subjects who have not been treated prior for diffuse, large B-cell, CD20-positive non-Hodgkin lymphoma.
Reference drug: Insulin glargine
Biosimilar(s)[19]
- Insulin glargine-yfgn (FDA-approved in July 2021)
Indications for biosimilar:
- Adult subjects with type 1 diabetes mellitus and type 2 diabetes mellitus
- Pediatric subjects with type 1 diabetes mellitus
Enhancing Healthcare Team Outcomes
As the development of biosimilars agents has progressed over the years, awareness in subspecialty practices encountering high incidence and prevalence of inflammatory diseases such as rheumatology, dermatology, and gastroenterology is also expanding.[4] Other specialties such as oncology and endocrinology have now also introduced biotherapeutics.[4] However, a gap in knowledge and confusion of biosimilars may exist in non-subspecialty practices, particularly with primary care clinicians (PCP). While PCP's are up to date on the more conventional original biologic treatments, many may not have gained familiarity and experience with the more current biosimilar innovative therapeutics, which in turn are more inexpensive, further increasing medication acquisition and compliance improving disease outcomes and the quality of life. [4]
The American journal of managed care (AJMC) discussed insights on biologics and biosimilars for inflammatory diseases. In the United States (US), fewer than 1% of dispensed prescribed medications consist of biological agents; however, the biologic agents value over one-fourth of all prescription costs in the US. The knowledge and awareness of biosimilars in specialty practices such as rheumatology and gastroenterology have increased in recent years due to the advocacy from respective specialty associations.[22] The preliminary approvals for biosimilars use for inflammatory diseases in medicine were issued for rheumatologic conditions. One of the gaps in knowledge for biosimilars use in medicine comes from when the FDA is making a governing action versus when a biosimilar is ultimately FDA-approved and available for use.
Biosimilars have been prevalent in Europe, Japan, and South Korea for over a decade, but their availability in the US was delayed due to the necessity of regulations dictating their pathway to market. The Biologics Price Competition and Innovation Act was implemented in 2009, and since the initial FDA approval was granted for biosimilar "filgrastim-sndz " in March 2015. To date, more biosimilars are being approved and marketed, with the latest approval coming on July 28, 2021, for biosimilar agent insulin glargine-yfgn. The proposed predictions for reducing costs vary from $54 billion to $250 billion within the midst of the 2020s onwards.[4][23]
For the cost-saving potential and improved patient access to agents for treating inflammatory diseases, recognition of biosimilars as safe and efficacious agents by patients, primary care clinicians, specialists, and other healthcare professionals is warranted to achieve market share.[23] The recognition of biosimilars as safe alternative agents to biologics may be subdued by education on the meticulous approval criteria required of biosimilars imposed by the FDA for prescribing clinicians and healthcare professionals.[4][24]
Several original biologics will be nearing closings of their patents, and prospective producers of biosimilars are anticipated and the importance of regulative criteria ensuring their safety and effectiveness in managing inflammatory diseases. More than 70% of biologic agents' costs were accumulated from biologics with lapsed patents, which would soon be off-patent.[4]
When producing and developing biosimilars, their primary structure and sequence of amino acids are identical to their respective reference products.[24] The minor variations may occur in the biosimilar tertiary or quarternary structure, with no meaningful clinical differences compared to the original biologics and treating inflammatory diseases.[4] Moreover, biosimilars also possess no discrepancies in safety, purity, efficacy, and potency compared to the original biologics.[4][24] Evidence-based impacts with biosimilars in clinical practice have been limited due to their legal matters and availability. Clinical and desired patient outcomes with biosimilars and choosing them over a conventional biologic for inflammatory diseases vary from specialty to specialty and on a case-by-case basis, depending on the individual and type of disease course.[24]
Some patients may have a more silent disease course, while others may be in remission. Clinicians may feel more comfortable with the standard of care and prescribe a biological agent due to successful patient outcomes.[24] Immunogenicity should also be considered when prescribing immunotherapeutics in a clinical setting, as it may also affect desired patient results.
When treating inflammatory diseases, a patient-centered approach should be regarded when considering the use of biosimilars. Biosimilars are deemed to be more affordable when compared to original biologics improving access to treatment options. When prescribing biosimilars, cost efficiency for the patient should be considered as it may only be more economical to the provider and payer but not so much to the patient.[22] As some subjects may be paying a certain amount of co-pay for conventional biologics, it is not assured the patients will receive a reduced rate if treated with a biosimilar.[22]
Patients may also feel comfortable continuing care with the original biologic initiated if they are already being treated compared to subjects with newly diagnosed inflammatory diseases, who may feel more open to a biosimilar.[24][22] An open-ended discussion and patient-centered approach between provider and patients regarding biosimilars, treatment options, and patient outcomes should be discussed.
Biosimilars ultimately demonstrate similarity to original biologics based on their characteristics of safety, efficacy, potency, tolerability but do not share similar cost considerations. Cost and prescribing considerations can ultimately make biosimilars the choice of biotherapeutics when prescribing biologics for inflammatory diseases.[24][22]
Barriers that need to be considered when prescribing biosimilars are patients not responding to the switch in medications or receiving no cost benefits due to their managed care plans.[23] The cost and prescribing considerations are a predominant focus of biosimilars in the US, as decreasing costs will improve patient access to biotherapeutics.[24][22] Cost variability for biosimilars may also affect their presence and use on the US market. Competition from other biopharmaceuticals such as price drops, incentives, and reimbursements from existing biologics may narrow the price gap between original biologics and biosimilars.[23]
Challenges clinicians may face when discussing biosimilars with patients for inflammatory diseases are medication switches in subjects with disease states already in remission. Patients may not want to risk replacing an already established agent that has demonstrated desired patient outcomes and disease stability with another agent similar to the primary medication for cost benefits.[24][22] Patient education and information on biosimilars are crucial for subjects already managed with biotherapeutics for inflammatory diseases. A better knowledge of biosimilars can overcome barriers in treatment.[22][4]
Clinician awareness for biosimilars use for inflammatory disease in clinical practice is gradually increasing through clinician programs through their respective specialty programs.[22] A greater understanding of the benefits, risks, similarities, safety, efficacy, costs, and prescribing considerations of biosimilars compared to the original biologics can aid clinicians to make informed decisions when utilizing and discussing biosimilars with their patients.[4][22]
The future for biosimilars use in inflammatory disease in the US is gaining positive traction.[22] To date, there are currently 30 FDA-approved biosimilars agents referenced from 10 original biologics since its very first approval in 2015. When looking at the current state of biosimilars in the European Union (EU), European Medicines Agency (EMA) has approved 65 biosimilars for the use of various diseases. The US FDA regulatory pathway of biosimilars is ordinarily alike of EMA with some variations.[25]
The EMA has served as a lead for setting regulatory pathways for biosimilars and their approvals as the first-ever biosimilar received approval in 2006 by the EMA.[25] Biosimilar agents approved for use in the EU include insulin, growth hormone, heparin/ low-molecular-weight heparin (LMWH), interferons, and monoclonal antibody products.[25][24][4] The FDA recently approved its first insulin biosimilar and first-ever interchangeable products. As advancements in technology and scientific methodology progress, biosimilars' development and regulatory conditions are also destined to improve.[25][4][23][24][26][22]
Nursing, Allied Health, and Interprofessional Team Interventions
Biosimilars are biological agents that are highly analogous to their reference products currently approved and licensed by the U.S. Food and Drug Administration (FDA). Biosimilars can be used in various inflammatory and autoimmune diseases such as rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, plaque psoriasis, inflammatory bowel disease: adult Crohn disease, ulcerative colitis, granulomatosis with polyangiitis (Wegener granulomatosis), microscopic and polyangiitis (MPA). With the latest advancements in biosimilars agents, it is now also indicated for use in specific malignancies, anemia, and diabetes mellitus—patients suffering from such conditions prompt care and oversight from an interprofessional team of healthcare professionals.
The interprofessional team should consist of a primary care clinician, specialists such as a rheumatologist, gastroenterologist, dermatologist, oncologist, and endocrinologist, alongside mid-level practitioners, nurses, a physical therapist (PT), and a pharmacist. Efficient care and communication within the interprofessional healthcare team concerning patients suffering from such ailments receiving biosimilars can improve patient access to biotherapeutics, resulting in clinical improvements, decreased disease progression, and a greater quality of life.
Prescribing clinicians and the healthcare team should be up to date with the latest guidelines, advancements, and latest FDA-approved indications for biosimilar use. Although FDA-approved biosimilars have extrapolated indications of their original biologic agents, some FDA labels of biosimilars do not designate all indications of their reference products and may potentially be considered off-label use.[4] Healthcare professionals should also review the difference between biosimilars and interchangeable biosimilars and thoroughly educate their patients being managed with such agents.[4]
Patients should be educated on biosimilars and their comparison to original biologics and reassured that FDA biosimilars possess no clinically meaningful differences in terms of their safety, purity, potency, or effectiveness compared to their respective reference biologic agents. The healthcare team should also advise patients about the potential adverse effects while receiving treatment as similar adverse effects are anticipated as the reference products. Immunogenicity should also be discussed when prescribing immunotherapeutics in a clinical setting, as it may also alter desired patient results. Additional educational material on biosimilars can be provided to patients from the FDA website.
Clinicians can expand their knowledge base and awareness of advances in biosimilars and their use in specialty practices from their respective specialty associations. Additional information can also be attained from the FDA website. The cost-saving potential of biosimilars should be taken into account and analyzed for the best treatment options for the patients. Biosimilars use in a clinical setting should be patient-focused and beneficial to the patient and not limited to provider and managed care plans. A thorough, open-ended discussion on biosimilars use for therapy between the interprofessional team, primarily the PCP and specialists, and their patients, is crucial as education on treatment with biotherapeutics in the management of inflammatory diseases can further increase medication trust and compliance and better clinician-patient rapport.