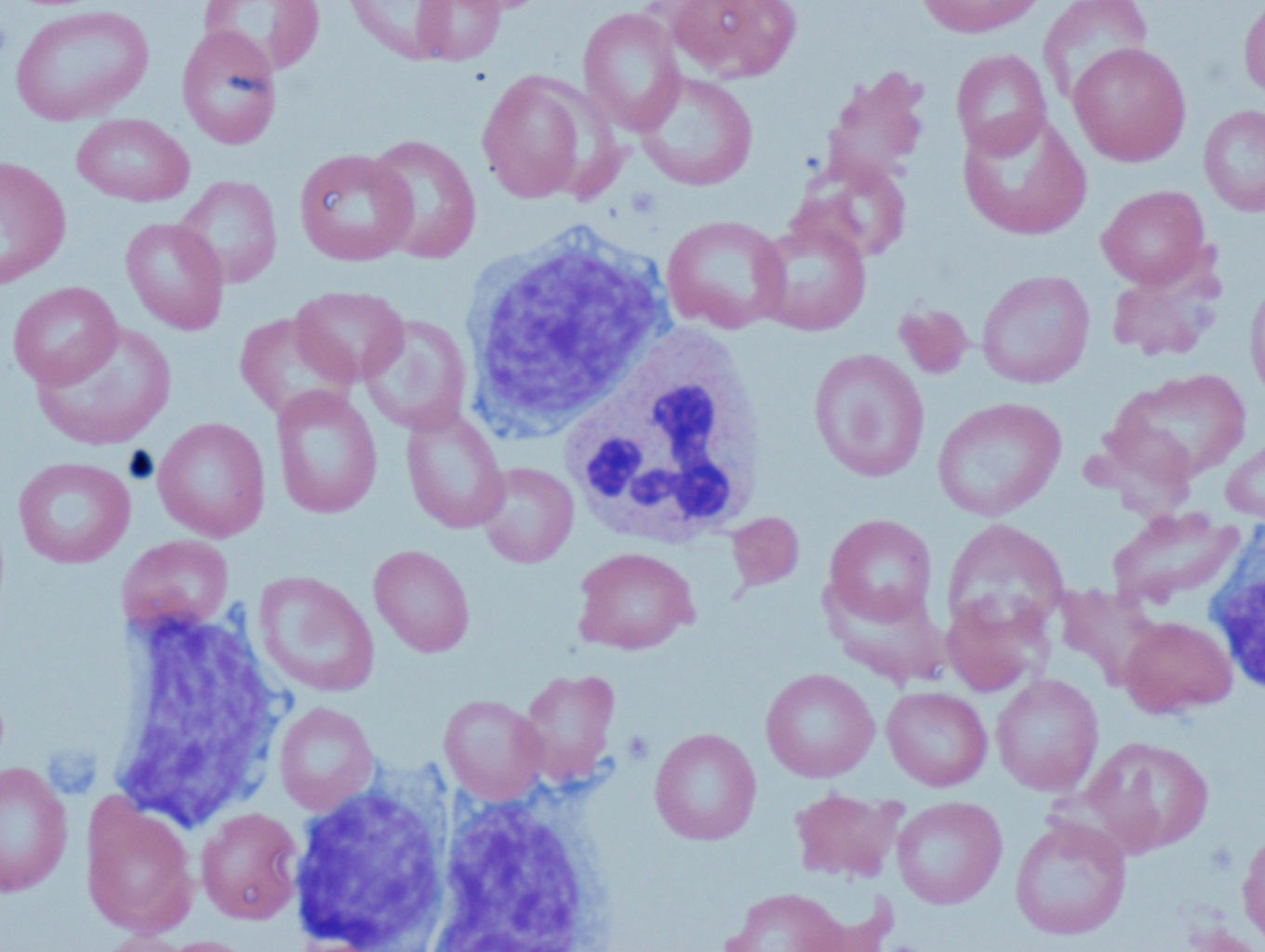
[1]
Brigden ML, Au S, Thompson S, Brigden S, Doyle P, Tsaparas Y. Infectious mononucleosis in an outpatient population: diagnostic utility of 2 automated hematology analyzers and the sensitivity and specificity of Hoagland's criteria in heterophile-positive patients. Archives of pathology & laboratory medicine. 1999 Oct:123(10):875-81
[PubMed PMID: 10506437]
[2]
Rosenberg ES, Caliendo AM, Walker BD. Acute HIV infection among patients tested for mononucleosis. The New England journal of medicine. 1999 Mar 25:340(12):969
[PubMed PMID: 10094651]
[3]
Fiala M, Heiner DC, Turner JA, Rosenbloom B, Guze LB. Infectious mononucleosis and mononucleosis syndromes. The Western journal of medicine. 1977 Jun:126(6):445-59
[PubMed PMID: 195404]
[4]
Arber DA, George TI. Bone marrow biopsy involvement by non-Hodgkin's lymphoma: frequency of lymphoma types, patterns, blood involvement, and discordance with other sites in 450 specimens. The American journal of surgical pathology. 2005 Dec:29(12):1549-57
[PubMed PMID: 16327427]
[5]
George TI. Malignant or benign leukocytosis. Hematology. American Society of Hematology. Education Program. 2012:2012():475-84. doi: 10.1182/asheducation-2012.1.475. Epub
[PubMed PMID: 23233622]
[6]
Matutes E, Parry-Jones N, Brito-Babapulle V, Wotherspoon A, Morilla R, Atkinson S, Elnenaei MO, Jain P, Giustolisi GM, A'Hern RP, Catovsky D. The leukemic presentation of mantle-cell lymphoma: disease features and prognostic factors in 58 patients. Leukemia & lymphoma. 2004 Oct:45(10):2007-15
[PubMed PMID: 15370245]
[7]
Woyach JA, Smucker K, Smith LL, Lozanski A, Zhong Y, Ruppert AS, Lucas D, Williams K, Zhao W, Rassenti L, Ghia E, Kipps TJ, Mantel R, Jones J, Flynn J, Maddocks K, O'Brien S, Furman RR, James DF, Clow F, Lozanski G, Johnson AJ, Byrd JC. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2014 Mar 20:123(12):1810-7. doi: 10.1182/blood-2013-09-527853. Epub 2014 Jan 10
[PubMed PMID: 24415539]
[8]
Snow AL, Xiao W, Stinson JR, Lu W, Chaigne-Delalande B, Zheng L, Pittaluga S, Matthews HF, Schmitz R, Jhavar S, Kuchen S, Kardava L, Wang W, Lamborn IT, Jing H, Raffeld M, Moir S, Fleisher TA, Staudt LM, Su HC, Lenardo MJ. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. The Journal of experimental medicine. 2012 Nov 19:209(12):2247-61. doi: 10.1084/jem.20120831. Epub 2012 Nov 5
[PubMed PMID: 23129749]
[9]
Troussard X, Cornet E, Lesesve JF, Kourel C, Mossafa H. Polyclonal B-cell lymphocytosis with binucleated lymphocytes (PPBL). OncoTargets and therapy. 2008 Oct 1:1():59-66
[PubMed PMID: 21127753]
[10]
Teggatz JR, Parkin J, Peterson L. Transient atypical lymphocytosis in patients with emergency medical conditions. Archives of pathology & laboratory medicine. 1987 Aug:111(8):712-4
[PubMed PMID: 3632284]
[11]
Heath CW Jr, Brodsky AL, Potolsky AI. Infectious mononucleosis in a general population. American journal of epidemiology. 1972 Jan:95(1):46-52
[PubMed PMID: 5007364]
[13]
Skoff TH, Hadler S, Hariri S. The Epidemiology of Nationally Reported Pertussis in the United States, 2000-2016. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2019 May 2:68(10):1634-1640. doi: 10.1093/cid/ciy757. Epub
[PubMed PMID: 30169627]
[14]
Rawstron AC, Green MJ, Kuzmicki A, Kennedy B, Fenton JA, Evans PA, O'Connor SJ, Richards SJ, Morgan GJ, Jack AS, Hillmen P. Monoclonal B lymphocytes with the characteristics of "indolent" chronic lymphocytic leukemia are present in 3.5% of adults with normal blood counts. Blood. 2002 Jul 15:100(2):635-9
[PubMed PMID: 12091358]
[15]
Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. The New England journal of medicine. 2004 Mar 25:350(13):1328-37
[PubMed PMID: 15044644]
[16]
Kubic VL, Kubic PT, Brunning RD. The morphologic and immunophenotypic assessment of the lymphocytosis accompanying Bordetella pertussis infection. American journal of clinical pathology. 1991 Jun:95(6):809-15
[PubMed PMID: 2042590]
[17]
Rezk E, Nofal YH, Hamzeh A, Aboujaib MF, AlKheder MA, Al Hammad MF. Steroids for symptom control in infectious mononucleosis. The Cochrane database of systematic reviews. 2015 Nov 8:2015(11):CD004402. doi: 10.1002/14651858.CD004402.pub3. Epub 2015 Nov 8
[PubMed PMID: 26558642]
Level 1 (high-level) evidence
[18]
Shanafelt TD, Wang XV, Kay NE, Hanson CA, O'Brien S, Barrientos J, Jelinek DF, Braggio E, Leis JF, Zhang CC, Coutre SE, Barr PM, Cashen AF, Mato AR, Singh AK, Mullane MP, Little RF, Erba H, Stone RM, Litzow M, Tallman M. Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. The New England journal of medicine. 2019 Aug 1:381(5):432-443. doi: 10.1056/NEJMoa1817073. Epub
[PubMed PMID: 31365801]
[19]
Buchwald DS, Rea TD, Katon WJ, Russo JE, Ashley RL. Acute infectious mononucleosis: characteristics of patients who report failure to recover. The American journal of medicine. 2000 Nov:109(7):531-7
[PubMed PMID: 11063953]
[20]
Teepe J, Broekhuizen BD, Ieven M, Loens K, Huygen K, Kretzschmar M, de Melker H, Butler CC, Little P, Stuart B, Coenen S, Goossens H, Verheij TJ, GRACE consortium. Prevalence, diagnosis, and disease course of pertussis in adults with acute cough: a prospective, observational study in primary care. The British journal of general practice : the journal of the Royal College of General Practitioners. 2015 Oct:65(639):e662-7. doi: 10.3399/bjgp15X686917. Epub
[PubMed PMID: 26412843]
Level 2 (mid-level) evidence
[21]
Cacoub P, Musette P, Descamps V, Meyer O, Speirs C, Finzi L, Roujeau JC. The DRESS syndrome: a literature review. The American journal of medicine. 2011 Jul:124(7):588-97. doi: 10.1016/j.amjmed.2011.01.017. Epub 2011 May 17
[PubMed PMID: 21592453]
[22]
Röllig C, Ehninger G. How I treat hyperleukocytosis in acute myeloid leukemia. Blood. 2015 May 21:125(21):3246-52. doi: 10.1182/blood-2014-10-551507. Epub 2015 Mar 16
[PubMed PMID: 25778528]
[23]
Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clinical advances in hematology & oncology : H&O. 2014 Jun:12(6):358-71
[PubMed PMID: 25003566]
Level 3 (low-level) evidence
[24]
Tsimberidou AM, Wen S, McLaughlin P, O'Brien S, Wierda WG, Lerner S, Strom S, Freireich EJ, Medeiros LJ, Kantarjian HM, Keating MJ. Other malignancies in chronic lymphocytic leukemia/small lymphocytic lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009 Feb 20:27(6):904-10. doi: 10.1200/JCO.2008.17.5398. Epub 2008 Dec 29
[PubMed PMID: 19114699]