Introduction
Genetic disorders traditionally fall into three main categories: single-gene defects, chromosomal abnormalities, and multifactorial conditions. A chromosomal abnormality, or chromosomal aberration, is a disorder characterized by a morphological or numerical alteration in single or multiple chromosomes, affecting autosomes, sex chromosomes, or both. The normal human karyotype contains approximately two meters of DNA organized into 46 chromosomes: 22 pairs of homologous autosomal chromosomes and a set of sex chromosomes that compromise two X chromosomes in females or an X and a Y chromosome in males. All the genetic necessary for growth and development derive from chromosomes (around 20 to 25 thousand genes). Chromosome abnormalities usually involve an error in cell division (mitosis or meiosis), which may occur in the prenatal, postnatal, or preimplantation periods. These alterations have significant clinical consequences, i.e., spontaneous abortions, stillbirths, neonatal death/hospitalization, malformations, intellectual disability, or an identifiable syndrome. Accurate identification of these chromosomal errors is essential for prevention strategies, genetic counseling, and appropriate treatment.
Development
Cytogenetics refers to the study of chromosomes. Although it has been known that chromosomes form the physical basis for inheritance for well over a hundred years, the discipline of mammalian cytogenetics, as we know it today, started around the mid-1950s.[1] In 1956, the diploid number of chromosomes in human cells was established at 46 instead of the previously accepted 48.[1][2] Additionally, a technique for chromosome preparation from leukocytes cultured from peripheral blood was accomplished by 1960.[3] From that moment, scientists could correctly describe the aberrations to the normal human chromosome number or structure.
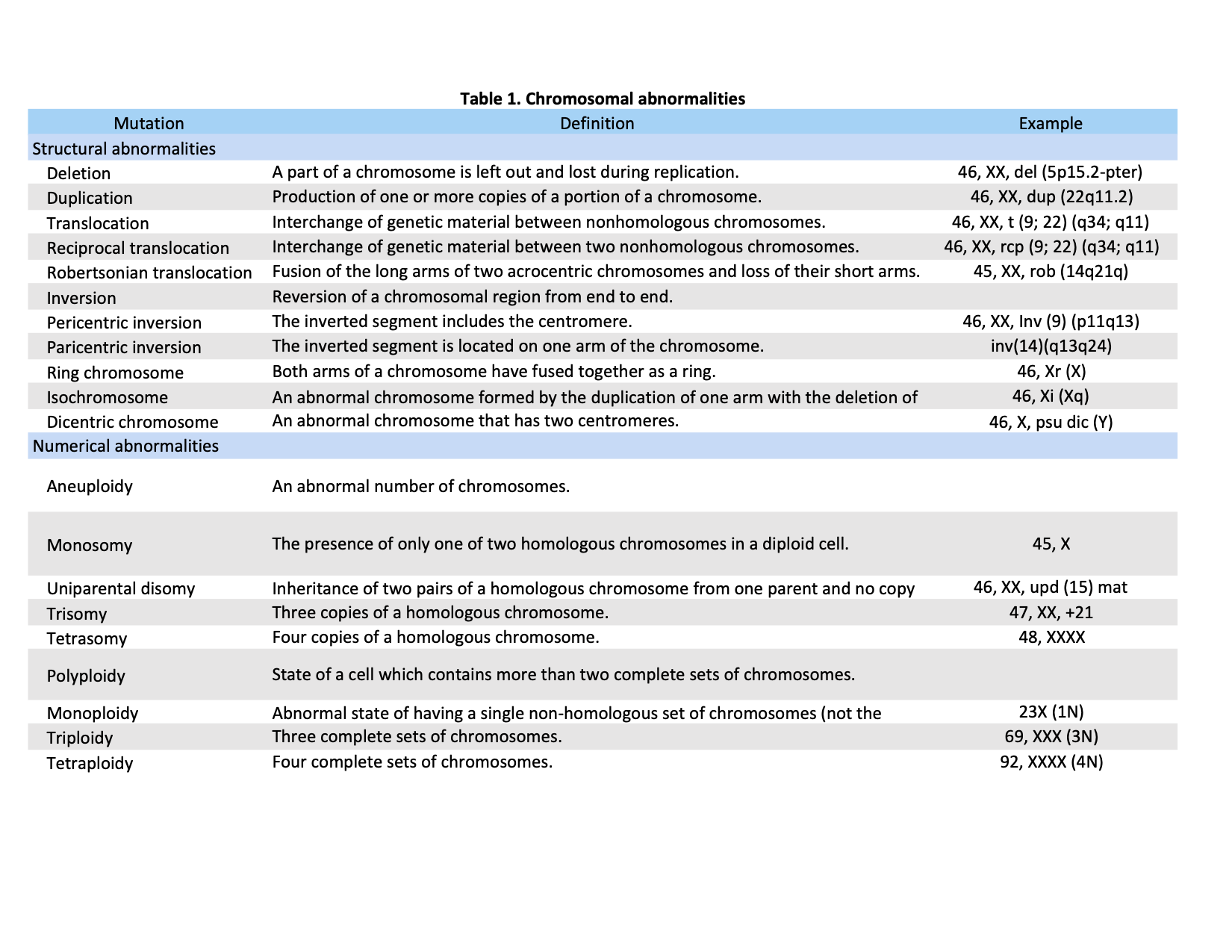
Chromosomal abnormalities generally fall into two basic categories: numerical or structural. Numerical disorders are considerably more common than structural ones; they involve any deviation from the normal diploid number for a given species. This category can further subdivide into a group in which individual chromosomes may be either missing or duplicated and a group in which whole haploid sets of chromosomes may be added or lost.[1] In contrast, structural abnormalities involve the genomic rearrangement of one or more chromosomes. Most structural aberrations result from the unequal exchange between chromosomes or enzymatic misrepair of two chromosome breakages.[4] Examples of this disorder include deletions, translocations, and inversions, duplications, ring chromosomes, and isochromosomes. Table 1 displays a list of definitions and examples of chromosomal abnormalities according to their classification (numerical or structural).
Chromosomal abnormalities may also classify as constitutional or acquired. Constitutional chromosomal abnormalities arise during gametogenesis or early embryogenesis and affect all or a major portion of an organism’s cells.[5] Their estimated incidence is around 20% to 50% of all human conceptions, and there is documentation of more than a thousand different anomalies in liveborn patients. Acquired chromosomal abnormalities typically develop during adulthood and affect a single clone of cells with a determined distribution in the body exclusively. These alterations are involved in the pathogenesis of many neoplasms, which will not be the focus of this review.[5]
Cellular
As previously stated, the normal human cell contains 46 chromosomes, except for enucleate cells (i.e., red blood cells), cell fragments (platelets), and haploid germline cells (eggs and sperm), which contain only 23 chromosomes.[6] Hence, numerical chromosome abnormalities may occur in exact multiples of the haploid number (n), called euploidy, or in every other case. Aneuploidy is a general condition used for an abnormal number of chromosomes consisting of one or more extra or missing chromosomes.[6] It is the most prevalent chromosome abnormality in humans (developing in 5 to 10% of all pregnancies) and is the leading genetic cause of miscarriage and congenital defects.[7][8] Most aneuploidies are lethal; nonetheless, there is a scarce amount of viable syndromes. Furthermore, polyploidy, the condition of a cell having more than two sets of all chromosomes, is not observed uncommonly in human embryos.[6] For example, triploidy (three sets of chromosomes or 3N) may occur in up to 3% of all human conceptions. Nevertheless, all polyploidies (monosomy, triploidy, tetraploidy, pentaploidy, hexaploidy, and so on) are not life compatible with humans.
Generally, an aneuploid chromosome set varies from the wild type by a small number of chromosomes (typically one). For example, trisomy (three copies of a specific homologous chromosome or 2N + 1) is the most frequent constitutional chromosomal abnormality in humans.[5] Research has identified an extra chromosome copy for every chromosome, but most of them are lethal during embryogenesis; even the known viable clinical syndromes for trisomies are more frequently observed in spontaneous abortions than in live births.[5][6] Moreover, monosomies (a single non-homologous set of chromosomes or 2N - 1) are remarkably rare in humans and practically limited to chromosomes 21 or X. These findings have led to the hypothesis that the consequences of such polyploidies are the product of an imbalance from critical gene products encoded on, what could only be, small gene-poor chromosomes.[5][6] Of note, numerical chromosomal abnormalities in sex chromosomes have different implications than in autosomes. For instance, aneuploidy in autosomes are less common than in sex chromosome, and trisomies seem to be tolerated better than monosomies.[1] This explains why having an extra or missing sex chromosome is more compatible with life and may have only milder consequences (trisomy) or equally severe consequences (monosomy) than autosome aneuploidies, as discussed later.
Regarding structural chromosomal abnormalities, there is one major mechanism of significant clinical importance called Robertsonian translocation. In this case, two acrocentric chromosomes break at the centromere, losing both of their short p arms and creating a new single chromosome containing one centromere and the q arms of both original chromosomes. Some individuals may have this kind of cytogenic aberration and appear phenotypically normal because only small amounts of genetic material are present in the short arms of acrocentric chromosomes. Thus, the two remaining long arms can compensate for this loss. However, in some cases, it can lead to genetic imbalances and the formation of zygotes with polyploidies (trisomies or monosomies) with significant phenotypical consequences (i.e., Down syndrome). The estimated carrier frequency for Robertsonian translocations in the general population is 1 in 1000.[4][6]
Molecular Level
Aneuploidy is most commonly the result of a failure to segregate chromosomes in cell division, meiosis I or II. This outcome includes non-disjunction, premature disjunction, or anaphase lag.[4] Non-disjunction is the most frequent phenomenon out of the previously mentioned; it occurs when the replicated chromosomes do not separate adequately during one of the two meiotic divisions, predominately arising during meiosis I of oocyte formation compared to sperm. If this error does occur, the resulting germs cells will either have an extra or a missing copy of a certain chromosome. In contrast, sex chromosome aneuploidies usually have a different origin: paternal meiotic non-disjunction is responsible for at least 50% of cases.[5] Different consequences occur depending on which meiotic cycle is involved in non-disjunction. In meiosis I, the gamete would have 24 chromosomes from both the paternal and the maternal homologs. In contrast, if non-disjunction occurs in meiosis II, the gamete would still have 24 chromosomes, but both copies would exclusively derive from either the paternal or maternal homolog.[4]
Several factors share links to human aneuploidies: mother’s age, the occurrence of a previous trisomy, aberrant recombination, and, although less strongly associated, apparently folic acid deficiency, obesity, smoking, and radiation may have some connection.[7] Specifically, the rates of trisomy 21 increase nearly exponentially from 35 years of maternal age (increasing in frequency from 10 to 20% to more than 60%).[6][9][10] On the other hand, if a new mutation occurs during embryogenesis or development (mitotic division), it may lead to a phenomenon termed mosaicism, two or more karyotypically unequivocally distinct cell lineages within a single embryo.[10]
Testing
In over a century in cytogenetics, there have been many new techniques developed for chromosome testing.[11] The two main methods used for chromosomal abnormality detection are karyotyping and fluorescent in-situ hybridization (FISH).
Karyotyping
Traditionally, karyotyping is considered the definitive method for detecting chromosomal abnormalities because of the accurate identification of fetal aneuploidies, structural rearrangements, and duplications and/or deletions of size greater than 5 Mb.[12] Karyotype refers to the entire set of chromosomes in an organism. In the process, cells require to be inoculated and cultured for 10 to 11 days at 37 °C, followed by the addition of colchicine solution, a mitosis blocker to arrest cells in metaphase. After incubation and centrifugation, the medium is replaced with a hypo-osmotic solution to produce cell lysis. Finally, the sample is fixed on a glass slide, and chromosomes are stained with a specific dye (i.e., Giemsa dye) for analysis under a light microscope. The diagnosis rates for chorionic villus sampling and amniocentesis are 97.8% and 99.4%, respectively.[12]
Unfortunately, this technique requires cells situated in the metaphase stage. Only 24% to 36% of embryos produce metaphase cells of sufficient quality for accurate quantification of chromosomes, and less than 25% of these cases are sufficient for complete cell analysis.[13] Additionally, as cells need to be cultured, the entire process usually takes about 1 to 2 weeks [6]. Nevertheless, in amniotic fluid samples, karyotyping is considered the “gold standard” for diagnosing fetal aneuploidies.[12]
Fluorescent in-situ Hybridization (FISH)
This method uses fluorescent-labeled DNA probes to assess the presence, location, and copy number of the complementary DNA sequence of interest in an individual’s genome by hybridization.[6][12] The probes must be labeled either directly or indirectly, depending on the presence of fluorescent nucleotides or nucleotides that act as haptens for fluorescently labeled antibodies, respectively. First, the sample is cultured for arrest in the metaphase stage. Following incubation, hybridization, and wash out to remove unbounded probes, the sample is viewed under a fluorescence microscope. This technique is particularly advantageous as a relatively rapid diagnostic tool for structural chromosomal abnormalities involving small segments of DNA (microdeletions, translocations, etc.) and for detecting trisomies due to a more distinct fluorescent reading.[2][11][12] Ultimately, FISH led to a diverse group of more powerful techniques such as spectral karyotyping (SKY), multicolor FISH (M-FISH), and comparative genomic hybridization (CGH), which overcome some initial limitations (single probe, color, and region of analysis).[2][12]
Finally, remember that some chromosome abnormalities can be detected with cytogenetic diagnostics but are not associated with clinical defects.[14]
Clinical Significance
Approximately 0.4 to 0.9% of newborns have chromosomal abnormalities, and about half of them have an abnormal phenotype.[5][9][15][16] The following paragraphs correlate the most clinically relevant aneuploidies and important key points on each:
Autosomal Trisomies
Trisomy 13 (Patau Syndrome)
- Incidence: Approximately 1 in 5000 to 1 in 16000 live births (third most common autosomal trisomy)
- Karyotype: 47, XX or XY, +13
- Clinical features: Microcephaly, holoprosencephaly, microphthalmia, small or poorly developed eyes (anophthalmia or cyclopia), cleft lip and palate, polydactyly, rocker-bottom feet, congenital heart disease, cryptorchidism, brain or spinal cord abnormalities, weak muscle tone at birth, severe intellectual disability.
- Prognosis: miscarriage, stillbirth, or early death (median survival around one year of age).[4][6]
Trisomy 18 (Edwards Syndrome)
- Incidence: Approximately 1/3000 to 1:5000 live births (second most common autosomal trisomy)
- Karyotype: 47, XX or XY, +18
- Clinical features: Low birth weight, microcephaly, micrognathia, low-set, malformed ears, clenched fists with overlapping fingers, congenital heart and renal abnormalities, rocker-bottom feet, severe intellectual disability, survival about one year.
- Prognosis: miscarriage, stillbirth, or early death (median survival around one year of age).[4][6]
Trisomy 21 (Down Syndrome)
- Incidence: Approximately 1:700 to 1/800 live births (most common autosomal trisomy)
- Karyotype: 47, XX or XY, +21
- Clinical features: Characteristic facial appearance, flat facies, prominent epicanthic folds, flat occiput, Brushfiel spot in irides, weak muscle tone at birth, single transverse palmar crease (simian crease), clinodactyly, congenital digestive and cardiac defects, intellectual disability, increased risk for leukemia, Alzheimer disease and hearing an vision problems
- Prognosis: Approximately 75% of conceptions die in embryonic or fetal life. Nevertheless, if birth occurs, the prognosis is relatively good (median survival is 47 years). It is the most common aneuploidy that is compatible with long-term survival.
- Note: Approximately 95% of patients with Down syndrome have trisomy 21. However, 4% of cases are due to Robertsonian translocation, and 1% is due to mosaicism.[4][6][17]
Sex Chromosome Aneuploidies
Klinefelter Syndrome
- Incidence: 1/500 to 1:1000 male births
- Karyotype: Typically 47, XXY (greater than 90%). However, other karyotypes have been described: 48, XXXY and 49, XXXXY and 46, XY/47, XXY (mosaicism)
- Clinical features: Increased height, long extremities, low upper/lower segment ratio, gynecomastia, reduced facial and body hair (female hair distribution), delayed and incomplete puberty, small testes (testicular atrophy), infertility, developmental delay (learning disabilities, delayed speech and language development), increased risk for breast cancer.
- Prognosis: Variable. It depends on the severity of clinical manifestations and treatment but overall is fairly good. The life span is slightly reduced.[4][6]
Triple X Syndrome
- Incidence: Approximately 1/1000 female births
- Karyotype: 47, XXX
- Clinical features: Presents with increased height and risk of learning disabilities, delayed development of speech, language, and motor skills, weak muscle tone, behavioral and emotional difficulties, seizures, kidney abnormalities
- Some cases may seem phenotypically normal.
- Prognosis: Variable. It depends on the severity of clinical manifestations and treatment but overall is fairly good.[4][6] Notes: In this aneuploidy, X chromosomes are inactivated as Barr bodies. Therefore, two extra Barr bodies are present at karyotyping.
XYY Syndrome
- Incidence: Approximately 1:1000 male births
- Karyotype: 47, XYY
- Clinical features: Increased height and risk of learning disabilities, delayed development of speech, language, and motor skills, weak muscle tone, hand tremors, seizures, asthma, scoliosis, behavioral and emotional difficulties
- As with triple X syndrome, some cases may seem phenotypically normal.
- Prognosis: Variable. It depends on the severity of clinical manifestations and treatment but overall is fairly good.[4][6]
Turner Syndrome
- Most common sex chromosomal abnormality in females and the most common genetic cause of primary amenorrhea
- Incidence: Approximately 1 in 2000 to 1 in 2500 live female births
- Karyotype: 45, X accounts for 45% of cases because most zygotes cannot survive extrauterine life. Remaining cases have a mosaic karyotype (45, X/46, XX, 45, X/ 46XY or 45, X/47, XXX) or a structural abnormality [isochromosome, 46, Xi (Xq) or (Xp), or ring chromosome, 46, Xr (X)]
- Clinical features: Short stature, webbed neck, low posterior hairline, shield chest, amenorrhea, absence of puberty, the early loss of ovarian function (ovarian dysgenesis), infertility, skeletal abnormalities (i.e., cubitus valgus), lymphedema, congenital kidney and/or heart disease (i.e., horseshoe kidney, coarctation of the aorta).
- Prognosis: Variable. It depends on the severity of clinical manifestations and treatment but overall is fairly good.[4][6][18]
Cytogenetic analysis of fetal cells required samples from amniotic fluid, chorionic villi, or fetal blood, which are sampled through invasive testing, such as amniocentesis, chorionic villus sampling, and cordocentesis, respectively.[12] These methods pose a risk of miscarriage and other serious complications. However, some noninvasive genetic prenatal tests have been developed to allow even higher detection rates of aneuploidies in high-risk pregnancies. These noninvasive tests analyze cell-free DNA (cfDNA) in the maternal serum, as nearly 3% to 15% of cfDNA in the maternal blood is of fetal origin.[19] The utilization of cfDNA has increased enormously since its introduction in 2011. Although these techniques are for screening purposes of common trisomies only and have limitations such as the possibility of test failure (2.6% to 5.4%), they pose significant advantages over other tests and probably will become widely used for prenatal screening in the future.[19] The detection of chromosomal aberrations early in pregnancy is crucial. Clinicians can cover and discuss several medical options with the parents to make the best-informed decision.[12]