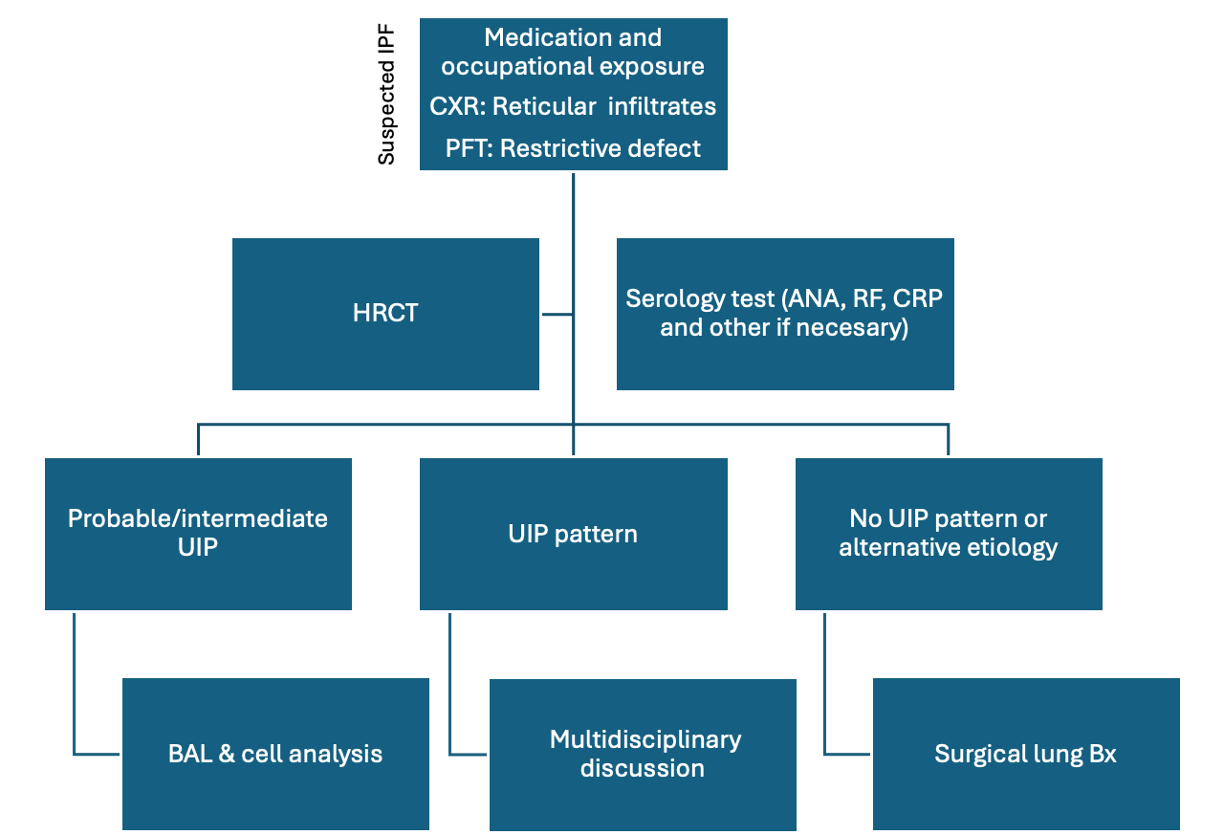
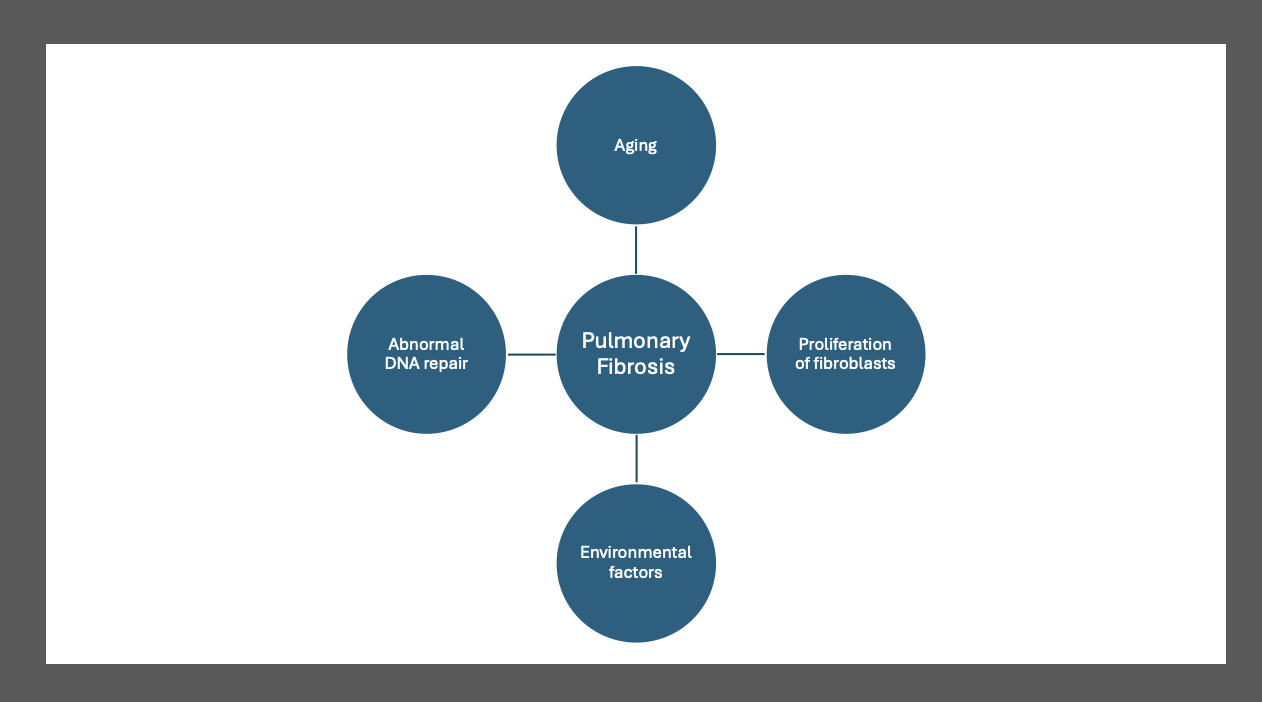
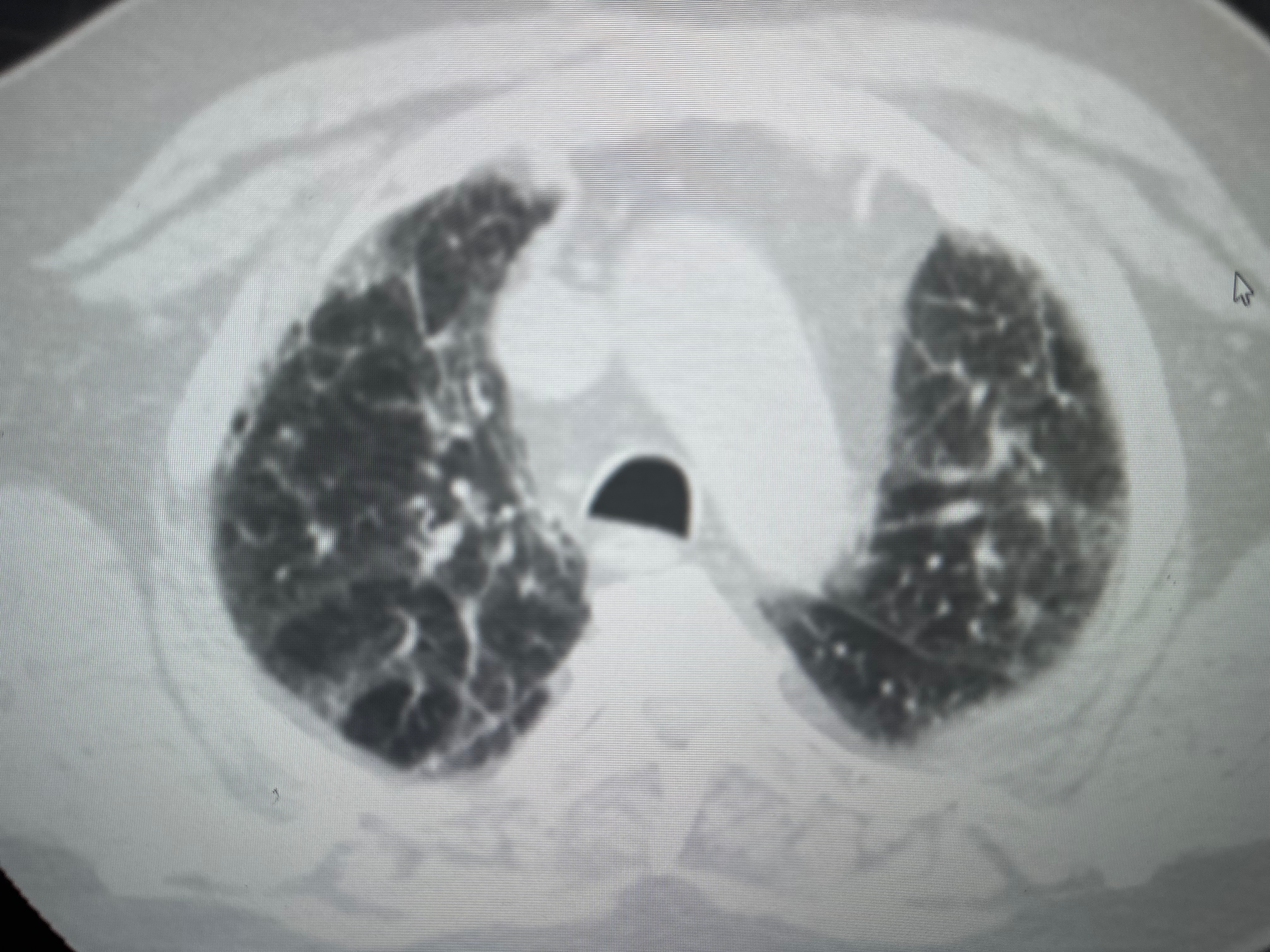
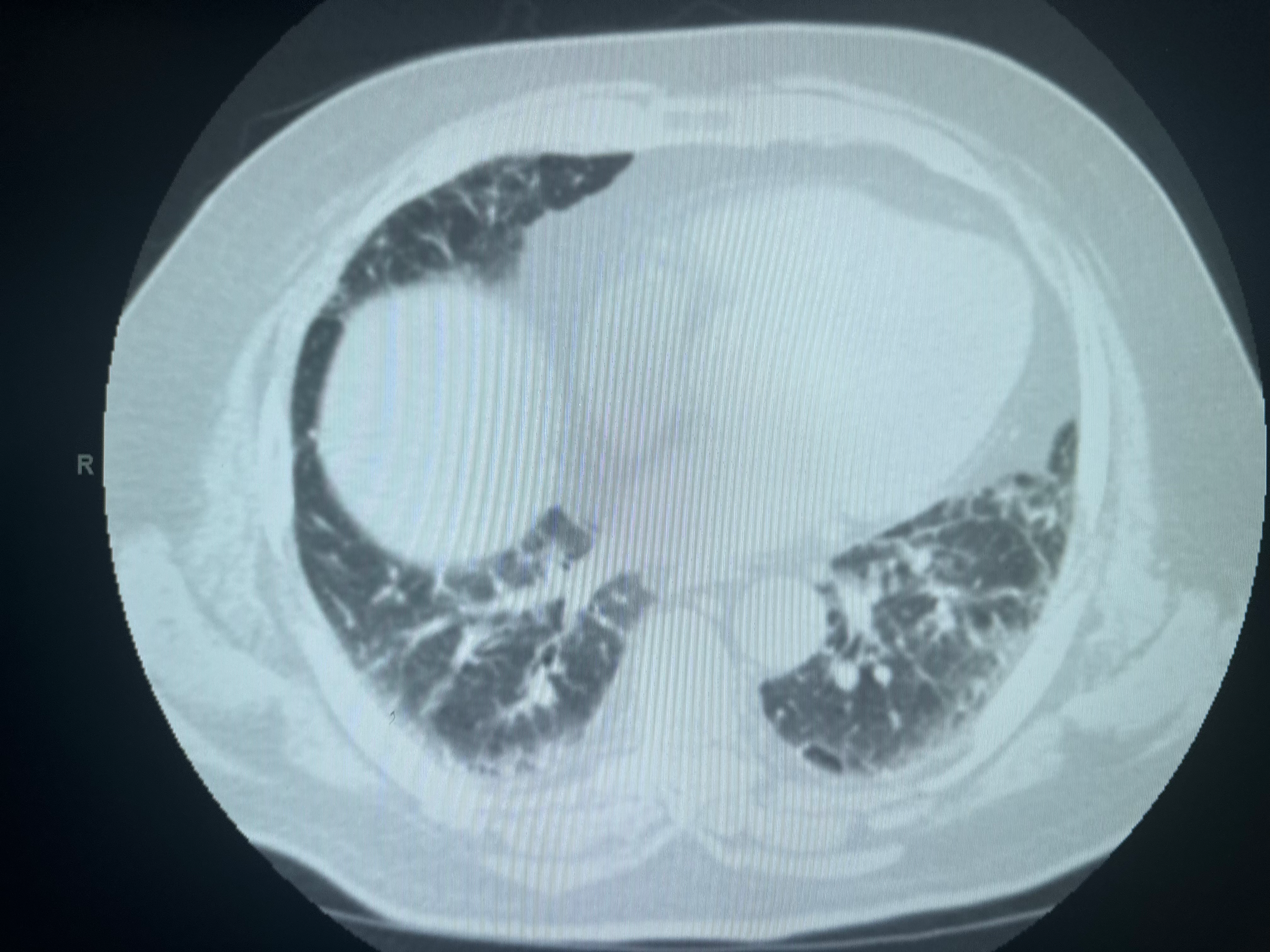
[1]
Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, Swigris JJ, Taniguchi H, Wells AU. Idiopathic pulmonary fibrosis. Nature reviews. Disease primers. 2017 Oct 20:3():17074. doi: 10.1038/nrdp.2017.74. Epub 2017 Oct 20
[PubMed PMID: 29052582]
[2]
Njock MS, Guiot J, Henket MA, Nivelles O, Thiry M, Dequiedt F, Corhay JL, Louis RE, Struman I. Sputum exosomes: promising biomarkers for idiopathic pulmonary fibrosis. Thorax. 2019 Mar:74(3):309-312. doi: 10.1136/thoraxjnl-2018-211897. Epub 2018 Sep 22
[PubMed PMID: 30244194]
[3]
Walsh SLF, Calandriello L, Silva M, Sverzellati N. Deep learning for classifying fibrotic lung disease on high-resolution computed tomography: a case-cohort study. The Lancet. Respiratory medicine. 2018 Nov:6(11):837-845. doi: 10.1016/S2213-2600(18)30286-8. Epub 2018 Sep 16
[PubMed PMID: 30232049]
Level 3 (low-level) evidence
[4]
Wells AU. Efficacy data in treatment extension studies of idiopathic pulmonary fibrosis: interpret with caution. The Lancet. Respiratory medicine. 2019 Jan:7(1):7-8. doi: 10.1016/S2213-2600(18)30385-0. Epub 2018 Sep 14
[PubMed PMID: 30224324]
[5]
Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F, Flaherty KR, Wells A, Martinez FJ, Azuma A, Bice TJ, Bouros D, Brown KK, Collard HR, Duggal A, Galvin L, Inoue Y, Jenkins RG, Johkoh T, Kazerooni EA, Kitaichi M, Knight SL, Mansour G, Nicholson AG, Pipavath SNJ, Buendía-Roldán I, Selman M, Travis WD, Walsh S, Wilson KC, American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2018 Sep 1:198(5):e44-e68. doi: 10.1164/rccm.201807-1255ST. Epub
[PubMed PMID: 30168753]
Level 1 (high-level) evidence
[6]
Mei Q, Liu Z, Zuo H, Yang Z, Qu J. Idiopathic Pulmonary Fibrosis: An Update on Pathogenesis. Frontiers in pharmacology. 2021:12():797292. doi: 10.3389/fphar.2021.797292. Epub 2022 Jan 19
[PubMed PMID: 35126134]
[7]
Günther A, Korfei M, Mahavadi P, von der Beck D, Ruppert C, Markart P. Unravelling the progressive pathophysiology of idiopathic pulmonary fibrosis. European respiratory review : an official journal of the European Respiratory Society. 2012 Jun 1:21(124):152-60. doi: 10.1183/09059180.00001012. Epub
[PubMed PMID: 22654088]
[8]
Heukels P, Moor CC, von der Thüsen JH, Wijsenbeek MS, Kool M. Inflammation and immunity in IPF pathogenesis and treatment. Respiratory medicine. 2019 Feb:147():79-91. doi: 10.1016/j.rmed.2018.12.015. Epub 2019 Jan 9
[PubMed PMID: 30704705]
[9]
Luppi F, Kalluri M, Faverio P, Kreuter M, Ferrara G. Idiopathic pulmonary fibrosis beyond the lung: understanding disease mechanisms to improve diagnosis and management. Respiratory research. 2021 Apr 17:22(1):109. doi: 10.1186/s12931-021-01711-1. Epub 2021 Apr 17
[PubMed PMID: 33865386]
Level 3 (low-level) evidence
[10]
Bano A, Chaker L, Muka T, Mattace-Raso FUS, Bally L, Franco OH, Peeters RP, Razvi S. Thyroid Function and the Risk of Fibrosis of the Liver, Heart, and Lung in Humans: A Systematic Review and Meta-Analysis. Thyroid : official journal of the American Thyroid Association. 2020 Jun:30(6):806-820. doi: 10.1089/thy.2019.0572. Epub 2020 Feb 13
[PubMed PMID: 31910097]
Level 1 (high-level) evidence
[11]
Zhang L, Wang Y, Wu G, Xiong W, Gu W, Wang CY. Macrophages: friend or foe in idiopathic pulmonary fibrosis? Respiratory research. 2018 Sep 6:19(1):170. doi: 10.1186/s12931-018-0864-2. Epub 2018 Sep 6
[PubMed PMID: 30189872]
[12]
Shioya M, Otsuka M, Yamada G, Umeda Y, Ikeda K, Nishikiori H, Kuronuma K, Chiba H, Takahashi H. Poorer Prognosis of Idiopathic Pleuroparenchymal Fibroelastosis Compared with Idiopathic Pulmonary Fibrosis in Advanced Stage. Canadian respiratory journal. 2018:2018():6043053. doi: 10.1155/2018/6043053. Epub 2018 Aug 13
[PubMed PMID: 30186537]
[13]
Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. The European respiratory journal. 2015 Sep:46(3):795-806. doi: 10.1183/09031936.00185114. Epub 2015 May 14
[PubMed PMID: 25976683]
Level 1 (high-level) evidence
[14]
Esposito DB, Lanes S, Donneyong M, Holick CN, Lasky JA, Lederer D, Nathan SD, O'Quinn S, Parker J, Tran TN. Idiopathic Pulmonary Fibrosis in United States Automated Claims. Incidence, Prevalence, and Algorithm Validation. American journal of respiratory and critical care medicine. 2015 Nov 15:192(10):1200-7. doi: 10.1164/rccm.201504-0818OC. Epub
[PubMed PMID: 26241562]
Level 1 (high-level) evidence
[15]
Olson AL, Gifford AH, Inase N, Fernández Pérez ER, Suda T. The epidemiology of idiopathic pulmonary fibrosis and interstitial lung diseases at risk of a progressive-fibrosing phenotype. European respiratory review : an official journal of the European Respiratory Society. 2018 Dec 31:27(150):. doi: 10.1183/16000617.0077-2018. Epub 2018 Dec 21
[PubMed PMID: 30578336]
[16]
Harari S, Madotto F, Caminati A, Conti S, Cesana G. Epidemiology of Idiopathic Pulmonary Fibrosis in Northern Italy. PloS one. 2016:11(2):e0147072. doi: 10.1371/journal.pone.0147072. Epub 2016 Feb 3
[PubMed PMID: 26841042]
[17]
Mostafaei S, Sayad B, Azar MEF, Doroudian M, Hadifar S, Behrouzi A, Riahi P, Hussen BM, Bayat B, Nahand JS, Moghoofei M. The role of viral and bacterial infections in the pathogenesis of IPF: a systematic review and meta-analysis. Respiratory research. 2021 Feb 12:22(1):53. doi: 10.1186/s12931-021-01650-x. Epub 2021 Feb 12
[PubMed PMID: 33579274]
Level 1 (high-level) evidence
[18]
Oldham JM, Collard HR. Comorbid Conditions in Idiopathic Pulmonary Fibrosis: Recognition and Management. Frontiers in medicine. 2017:4():123. doi: 10.3389/fmed.2017.00123. Epub 2017 Aug 2
[PubMed PMID: 28824912]
[19]
Papadogiannis G, Bouloukaki I, Mermigkis C, Michelakis S, Ermidou C, Mauroudi E, Moniaki V, Tzanakis N, Antoniou KM, Schiza SE. Patients with idiopathic pulmonary fibrosis with and without obstructive sleep apnea: differences in clinical characteristics, clinical outcomes, and the effect of PAP treatment. Journal of clinical sleep medicine : JCSM : official publication of the American Academy of Sleep Medicine. 2021 Mar 1:17(3):533-544. doi: 10.5664/jcsm.8932. Epub
[PubMed PMID: 33108270]
Level 2 (mid-level) evidence
[20]
Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, Aso H, Sakamoto K, Hasegawa Y. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration; international review of thoracic diseases. 2013:85(6):456-63. doi: 10.1159/000345221. Epub 2012 Dec 19
[PubMed PMID: 23257350]
[21]
Izbicki G, Ben-Dor I, Shitrit D, Bendayan D, Aldrich TK, Kornowski R, Kramer MR. The prevalence of coronary artery disease in end-stage pulmonary disease: is pulmonary fibrosis a risk factor? Respiratory medicine. 2009 Sep:103(9):1346-9. doi: 10.1016/j.rmed.2009.03.012. Epub 2009 Apr 11
[PubMed PMID: 19362458]
[22]
Lee JH, Lee HH, Park HJ, Kim S, Kim YJ, Lee JS, Kim HC. Venous thromboembolism in patients with idiopathic pulmonary fibrosis, based on nationwide claim data. Therapeutic advances in respiratory disease. 2023 Jan-Dec:17():17534666231155772. doi: 10.1177/17534666231155772. Epub
[PubMed PMID: 36846942]
Level 3 (low-level) evidence
[23]
Lee JS, Ryu JH, Elicker BM, Lydell CP, Jones KD, Wolters PJ, King TE Jr, Collard HR. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine. 2011 Dec 15:184(12):1390-4. doi: 10.1164/rccm.201101-0138OC. Epub 2011 Jun 23
[PubMed PMID: 21700909]
[24]
Kreuter M, Wuyts W, Renzoni E, Koschel D, Maher TM, Kolb M, Weycker D, Spagnolo P, Kirchgaessler KU, Herth FJ, Costabel U. Antacid therapy and disease outcomes in idiopathic pulmonary fibrosis: a pooled analysis. The Lancet. Respiratory medicine. 2016 May:4(5):381-9. doi: 10.1016/S2213-2600(16)00067-9. Epub 2016 Mar 31
[PubMed PMID: 27050871]
[25]
Linden PA, Gilbert RJ, Yeap BY, Boyle K, Deykin A, Jaklitsch MT, Sugarbaker DJ, Bueno R. Laparoscopic fundoplication in patients with end-stage lung disease awaiting transplantation. The Journal of thoracic and cardiovascular surgery. 2006 Feb:131(2):438-46
[PubMed PMID: 16434276]
[26]
Oldham JM, Kumar D, Lee C, Patel SB, Takahashi-Manns S, Demchuk C, Strek ME, Noth I. Thyroid Disease Is Prevalent and Predicts Survival in Patients With Idiopathic Pulmonary Fibrosis. Chest. 2015 Sep:148(3):692-700. doi: 10.1378/chest.14-2714. Epub
[PubMed PMID: 25811599]
[27]
Romagnoli M, Colby TV, Berthet JP, Gamez AS, Mallet JP, Serre I, Cancellieri A, Cavazza A, Solovei L, Dell'Amore A, Dolci G, Guerrieri A, Reynaud P, Bommart S, Zompatori M, Dalpiaz G, Nava S, Trisolini R, Suehs CM, Vachier I, Molinari N, Bourdin A. Poor Concordance between Sequential Transbronchial Lung Cryobiopsy and Surgical Lung Biopsy in the Diagnosis of Diffuse Interstitial Lung Diseases. American journal of respiratory and critical care medicine. 2019 May 15:199(10):1249-1256. doi: 10.1164/rccm.201810-1947OC. Epub
[PubMed PMID: 30864813]
[28]
Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annual review of pathology. 2014:9():157-79. doi: 10.1146/annurev-pathol-012513-104706. Epub 2013 Sep 13
[PubMed PMID: 24050627]
[29]
Mukhopadhyay S. Usual interstitial pneumonia (UIP): a clinically significant pathologic diagnosis. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2022 May:35(5):580-588. doi: 10.1038/s41379-022-01053-3. Epub 2022 Feb 28
[PubMed PMID: 35228663]
[30]
Chambers DC, Clarke BE, McGaughran J, Garcia CK. Lung fibrosis, premature graying, and macrocytosis. American journal of respiratory and critical care medicine. 2012 Sep 1:186(5):e8-9. doi: 10.1164/rccm.201112-2175IM. Epub
[PubMed PMID: 22942352]
[31]
Dai J, Cai H, Zhuang Y, Wu Y, Min H, Li J, Shi Y, Gao Q, Yi L. Telomerase gene mutations and telomere length shortening in patients with idiopathic pulmonary fibrosis in a Chinese population. Respirology (Carlton, Vic.). 2015 Jan:20(1):122-8. doi: 10.1111/resp.12422. Epub 2014 Oct 23
[PubMed PMID: 25346280]
[32]
Wytrychowski K, Hans-Wytrychowska A, Nowakowska B. Familial idiopathic pulmonary fibrosis. Advances in experimental medicine and biology. 2013:788():363-7. doi: 10.1007/978-94-007-6627-3_49. Epub
[PubMed PMID: 23835999]
Level 3 (low-level) evidence
[33]
Guenther A, Krauss E, Tello S, Wagner J, Paul B, Kuhn S, Maurer O, Heinemann S, Costabel U, Barbero MAN, Müller V, Bonniaud P, Vancheri C, Wells A, Vasakova M, Pesci A, Sofia M, Klepetko W, Seeger W, Drakopanagiotakis F, Crestani B. The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respiratory research. 2018 Jul 28:19(1):141. doi: 10.1186/s12931-018-0845-5. Epub 2018 Jul 28
[PubMed PMID: 30055613]
[35]
Nakatsuka Y, Handa T, Kokosi M, Tanizawa K, Puglisi S, Jacob J, Sokai A, Ikezoe K, Kanatani KT, Kubo T, Tomioka H, Taguchi Y, Nagai S, Chin K, Mishima M, Wells AU, Hirai T. The Clinical Significance of Body Weight Loss in Idiopathic Pulmonary Fibrosis Patients. Respiration; international review of thoracic diseases. 2018:96(4):338-347. doi: 10.1159/000490355. Epub 2018 Aug 21
[PubMed PMID: 30130749]
[36]
Walsh SLF. Imaging biomarkers and staging in IPF. Current opinion in pulmonary medicine. 2018 Sep:24(5):445-452. doi: 10.1097/MCP.0000000000000507. Epub
[PubMed PMID: 30015679]
Level 3 (low-level) evidence
[37]
Mangaonkar AA, Patnaik MM. Short Telomere Syndromes in Clinical Practice: Bridging Bench and Bedside. Mayo Clinic proceedings. 2018 Jul:93(7):904-916. doi: 10.1016/j.mayocp.2018.03.020. Epub 2018 May 24
[PubMed PMID: 29804726]
[38]
National Lung Screening Trial Research Team, Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD, Fagerstrom RM, Gareen IF, Gatsonis C, Marcus PM, Sicks JD. Reduced lung-cancer mortality with low-dose computed tomographic screening. The New England journal of medicine. 2011 Aug 4:365(5):395-409. doi: 10.1056/NEJMoa1102873. Epub 2011 Jun 29
[PubMed PMID: 21714641]
[39]
Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, Kreuter M, Lynch DA, Maher TM, Martinez FJ, Molina-Molina M, Myers JL, Nicholson AG, Ryerson CJ, Strek ME, Troy LK, Wijsenbeek M, Mammen MJ, Hossain T, Bissell BD, Herman DD, Hon SM, Kheir F, Khor YH, Macrea M, Antoniou KM, Bouros D, Buendia-Roldan I, Caro F, Crestani B, Ho L, Morisset J, Olson AL, Podolanczuk A, Poletti V, Selman M, Ewing T, Jones S, Knight SL, Ghazipura M, Wilson KC. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2022 May 1:205(9):e18-e47. doi: 10.1164/rccm.202202-0399ST. Epub
[PubMed PMID: 35486072]
Level 1 (high-level) evidence
[40]
Lentz RJ, Argento AC, Colby TV, Rickman OB, Maldonado F. Transbronchial cryobiopsy for diffuse parenchymal lung disease: a state-of-the-art review of procedural techniques, current evidence, and future challenges. Journal of thoracic disease. 2017 Jul:9(7):2186-2203. doi: 10.21037/jtd.2017.06.96. Epub
[PubMed PMID: 28840020]
[41]
Ravaglia C, Wells AU, Tomassetti S, Gurioli C, Gurioli C, Dubini A, Cavazza A, Colby TV, Piciucchi S, Puglisi S, Bosi M, Poletti V. Diagnostic yield and risk/benefit analysis of trans-bronchial lung cryobiopsy in diffuse parenchymal lung diseases: a large cohort of 699 patients. BMC pulmonary medicine. 2019 Jan 16:19(1):16. doi: 10.1186/s12890-019-0780-3. Epub 2019 Jan 16
[PubMed PMID: 30651103]
[42]
Furini F, Carnevale A, Casoni GL, Guerrini G, Cavagna L, Govoni M, Sciré CA. The Role of the Multidisciplinary Evaluation of Interstitial Lung Diseases: Systematic Literature Review of the Current Evidence and Future Perspectives. Frontiers in medicine. 2019:6():246. doi: 10.3389/fmed.2019.00246. Epub 2019 Oct 31
[PubMed PMID: 31750308]
Level 1 (high-level) evidence
[43]
Cerri S, Monari M, Guerrieri A, Donatelli P, Bassi I, Garuti M, Luppi F, Betti S, Bandelli G, Carpano M, Bacchi Reggiani ML, Tonelli R, Clini E, Nava S. Real-life comparison of pirfenidone and nintedanib in patients with idiopathic pulmonary fibrosis: A 24-month assessment. Respiratory medicine. 2019 Nov:159():105803. doi: 10.1016/j.rmed.2019.105803. Epub 2019 Oct 18
[PubMed PMID: 31670147]
[44]
Homma S, Bando M, Azuma A, Sakamoto S, Sugino K, Ishii Y, Izumi S, Inase N, Inoue Y, Ebina M, Ogura T, Kishi K, Kishaba T, Kido T, Gemma A, Goto Y, Sasaki S, Johkoh T, Suda T, Takahashi K, Takahashi H, Taguchi Y, Date H, Taniguchi H, Nakayama T, Nishioka Y, Hasegawa Y, Hattori N, Fukuoka J, Miyamoto A, Mukae H, Yokoyama A, Yoshino I, Watanabe K, Ministry of Health, Labour and Welfare, the Study Group on Diffuse Pulmonary Disorders, Scientific Research/Research on Intractable Diseases, and Japanese Respiratory Society. Japanese guideline for the treatment of idiopathic pulmonary fibrosis. Respiratory investigation. 2018 Jul:56(4):268-291. doi: 10.1016/j.resinv.2018.03.003. Epub 2018 Jul 3
[PubMed PMID: 29980444]
[45]
Cheng L, Tan B, Yin Y, Wang S, Jia L, Warner G, Jia G, Jiang W. Short- and long-term effects of pulmonary rehabilitation for idiopathic pulmonary fibrosis: a systematic review and meta-analysis. Clinical rehabilitation. 2018 Oct:32(10):1299-1307. doi: 10.1177/0269215518779122. Epub 2018 May 30
[PubMed PMID: 29843523]
Level 1 (high-level) evidence
[46]
Tolle LB, Southern BD, Culver DA, Horowitz JC. Idiopathic pulmonary fibrosis: What primary care physicians need to know. Cleveland Clinic journal of medicine. 2018 May:85(5):377-386. doi: 10.3949/ccjm.85a.17018. Epub
[PubMed PMID: 29733782]
[47]
Harari S, Wells AU, Wuyts WA, Nathan SD, Kirchgaessler KU, Bengus M, Behr J. The 6-min walk test as a primary end-point in interstitial lung disease. European respiratory review : an official journal of the European Respiratory Society. 2022 Sep 30:31(165):. doi: 10.1183/16000617.0087-2022. Epub 2022 Aug 23
[PubMed PMID: 36002171]
[48]
Johannson KA, Pendharkar SR, Mathison K, Fell CD, Guenette JA, Kalluri M, Kolb M, Ryerson CJ. Supplemental Oxygen in Interstitial Lung Disease: An Art in Need of Science. Annals of the American Thoracic Society. 2017 Sep:14(9):1373-1377. doi: 10.1513/AnnalsATS.201702-137OI. Epub
[PubMed PMID: 28644693]
[49]
Long-Term Oxygen Treatment Trial Research Group, Albert RK, Au DH, Blackford AL, Casaburi R, Cooper JA Jr, Criner GJ, Diaz P, Fuhlbrigge AL, Gay SE, Kanner RE, MacIntyre N, Martinez FJ, Panos RJ, Piantadosi S, Sciurba F, Shade D, Stibolt T, Stoller JK, Wise R, Yusen RD, Tonascia J, Sternberg AL, Bailey W. A Randomized Trial of Long-Term Oxygen for COPD with Moderate Desaturation. The New England journal of medicine. 2016 Oct 27:375(17):1617-1627
[PubMed PMID: 27783918]
Level 1 (high-level) evidence
[50]
Dowman L, Hill CJ, May A, Holland AE. Pulmonary rehabilitation for interstitial lung disease. The Cochrane database of systematic reviews. 2021 Feb 1:2(2):CD006322. doi: 10.1002/14651858.CD006322.pub4. Epub 2021 Feb 1
[PubMed PMID: 34559419]
Level 1 (high-level) evidence
[51]
Nolan CM, Polgar O, Schofield SJ, Patel S, Barker RE, Walsh JA, Ingram KA, George PM, Molyneaux PL, Maher TM, Man WD. Pulmonary Rehabilitation in Idiopathic Pulmonary Fibrosis and COPD: A Propensity-Matched Real-World Study. Chest. 2022 Mar:161(3):728-737. doi: 10.1016/j.chest.2021.10.021. Epub 2021 Oct 23
[PubMed PMID: 34699771]
[52]
Marijic P, Schwarzkopf L, Maier W, Trudzinski F, Schwettmann L, Kreuter M. Effects of Influenza Vaccination in Patients with Interstitial Lung Diseases: An Epidemiological Claims Data Analysis. Annals of the American Thoracic Society. 2022 Sep:19(9):1479-1488. doi: 10.1513/AnnalsATS.202112-1359OC. Epub
[PubMed PMID: 35312465]
Level 2 (mid-level) evidence
[53]
Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. The New England journal of medicine. 2012 May 24:366(21):1968-77. doi: 10.1056/NEJMoa1113354. Epub 2012 May 20
[PubMed PMID: 22607134]
[54]
Petnak T, Lertjitbanjong P, Thongprayoon C, Moua T. Impact of Antifibrotic Therapy on Mortality and Acute Exacerbation in Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. Chest. 2021 Nov:160(5):1751-1763. doi: 10.1016/j.chest.2021.06.049. Epub 2021 Jul 2
[PubMed PMID: 34217681]
Level 1 (high-level) evidence
[55]
Harrison SA, Bedossa P, Guy CD, Schattenberg JM, Loomba R, Taub R, Labriola D, Moussa SE, Neff GW, Rinella ME, Anstee QM, Abdelmalek MF, Younossi Z, Baum SJ, Francque S, Charlton MR, Newsome PN, Lanthier N, Schiefke I, Mangia A, Pericàs JM, Patil R, Sanyal AJ, Noureddin M, Bansal MB, Alkhouri N, Castera L, Rudraraju M, Ratziu V, MAESTRO-NASH Investigators. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. The New England journal of medicine. 2024 Feb 8:390(6):497-509. doi: 10.1056/NEJMoa2309000. Epub
[PubMed PMID: 38324483]
Level 1 (high-level) evidence
[56]
Costabel U, Albera C, Glassberg MK, Lancaster LH, Wuyts WA, Petzinger U, Gilberg F, Kirchgaessler KU, Noble PW. Effect of pirfenidone in patients with more advanced idiopathic pulmonary fibrosis. Respiratory research. 2019 Mar 12:20(1):55. doi: 10.1186/s12931-019-1021-2. Epub 2019 Mar 12
[PubMed PMID: 30866942]
[57]
Mandovra NP, Vaidya PJ, Shah RS, Nighojkar AS, Chavhan VB, Lohiya A, Leuppi JD, Leuppi-Taegtmeyer A, Chhajed PN. Factors Affecting Best-Tolerated Dose of Pirfenidone in Patients with Fibrosing Interstitial Lung Disease. Journal of clinical medicine. 2023 Oct 13:12(20):. doi: 10.3390/jcm12206513. Epub 2023 Oct 13
[PubMed PMID: 37892651]
[58]
Xaubet A, Serrano-Mollar A, Ancochea J. Pirfenidone for the treatment of idiopathic pulmonary fibrosis. Expert opinion on pharmacotherapy. 2014 Feb:15(2):275-81. doi: 10.1517/14656566.2014.867328. Epub 2013 Dec 6
[PubMed PMID: 24308635]
Level 3 (low-level) evidence
[59]
Wind S, Schmid U, Freiwald M, Marzin K, Lotz R, Ebner T, Stopfer P, Dallinger C. Clinical Pharmacokinetics and Pharmacodynamics of Nintedanib. Clinical pharmacokinetics. 2019 Sep:58(9):1131-1147. doi: 10.1007/s40262-019-00766-0. Epub
[PubMed PMID: 31016670]
[60]
Maher TM, Strek ME. Antifibrotic therapy for idiopathic pulmonary fibrosis: time to treat. Respiratory research. 2019 Sep 6:20(1):205. doi: 10.1186/s12931-019-1161-4. Epub 2019 Sep 6
[PubMed PMID: 31492155]
[61]
Pitre T, Khalid MF, Cui S, Zhang MC, Husnudinov R, Mah J, Helmczi W, Su J, Guy B, Scallan C, Jones A, Zeraatkar D. Sildenafil for idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Pulmonary pharmacology & therapeutics. 2022 Jun:73-74():102128. doi: 10.1016/j.pupt.2022.102128. Epub 2022 Apr 20
[PubMed PMID: 35452834]
Level 1 (high-level) evidence
[62]
Somogyi V, Chaudhuri N, Torrisi SE, Kahn N, Müller V, Kreuter M. The therapy of idiopathic pulmonary fibrosis: what is next? European respiratory review : an official journal of the European Respiratory Society. 2019 Sep 30:28(153):. doi: 10.1183/16000617.0021-2019. Epub 2019 Sep 4
[PubMed PMID: 31484664]
[63]
Chung JH, Oldham JM, Montner SM, Vij R, Adegunsoye A, Husain AN, Noth I, Lynch DA, Strek ME. CT-Pathologic Correlation of Major Types of Pulmonary Fibrosis: Insights for Revisions to Current Guidelines. AJR. American journal of roentgenology. 2018 May:210(5):1034-1041. doi: 10.2214/AJR.17.18947. Epub 2018 Mar 16
[PubMed PMID: 29547052]
[64]
Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, Brozek JL, Collard HR, Cunningham W, Homma S, Johkoh T, Martinez FJ, Myers J, Protzko SL, Richeldi L, Rind D, Selman M, Theodore A, Wells AU, Hoogsteden H, Schünemann HJ, American Thoracic Society, European Respiratory society, Japanese Respiratory Society, Latin American Thoracic Association. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. American journal of respiratory and critical care medicine. 2015 Jul 15:192(2):e3-19. doi: 10.1164/rccm.201506-1063ST. Epub
[PubMed PMID: 26177183]
Level 1 (high-level) evidence
[65]
Micco A, Carpentieri E, Di Sorbo A, Chetta A, Del Donno M. Palliative care and end of life management in patients with idiopathic pulmonary fibrosis. Multidisciplinary respiratory medicine. 2023 Jan 17:18():896. doi: 10.4081/mrm.2023.896. Epub 2023 Feb 21
[PubMed PMID: 36909932]
[66]
Rush B, Wiskar K, Berger L, Griesdale D. The use of mechanical ventilation in patients with idiopathic pulmonary fibrosis in the United States: A nationwide retrospective cohort analysis. Respiratory medicine. 2016 Feb:111():72-6. doi: 10.1016/j.rmed.2015.12.005. Epub 2015 Dec 21
[PubMed PMID: 26733227]
Level 2 (mid-level) evidence
[67]
Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, Srivastava A, de Castro JPW, DeIuliis G, Ahangari F, Woolard T, Aurelien N, Arrojo E Drigo R, Gan Y, Graham M, Liu X, Homer RJ, Scanlan TS, Mannam P, Lee PJ, Herzog EL, Bianco AC, Kaminski N. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nature medicine. 2018 Jan:24(1):39-49. doi: 10.1038/nm.4447. Epub 2017 Dec 4
[PubMed PMID: 29200204]
[68]
Zheng Q, Cox IA, Campbell JA, Xia Q, Otahal P, de Graaff B, Corte TJ, Teoh AKY, Walters EH, Palmer AJ. Mortality and survival in idiopathic pulmonary fibrosis: a systematic review and meta-analysis. ERJ open research. 2022 Jan:8(1):. pii: 00591-2021. doi: 10.1183/23120541.00591-2021. Epub 2022 Mar 14
[PubMed PMID: 35295232]
Level 1 (high-level) evidence
[69]
Serajeddini H, Rogliani P, Mura M. Multi-dimensional Assessment of IPF Across a Wide Range of Disease Severity. Lung. 2018 Dec:196(6):707-713. doi: 10.1007/s00408-018-0152-4. Epub 2018 Aug 27
[PubMed PMID: 30151723]
[70]
Barratt SL, Creamer A, Hayton C, Chaudhuri N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. Journal of clinical medicine. 2018 Aug 6:7(8):. doi: 10.3390/jcm7080201. Epub 2018 Aug 6
[PubMed PMID: 30082599]
Level 3 (low-level) evidence
[71]
Jeong SO, Uh ST, Park S, Kim HS. Effects of patient satisfaction and confidence on the success of treatment of combined rheumatic disease and interstitial lung disease in a multidisciplinary outpatient clinic. International journal of rheumatic diseases. 2018 Aug:21(8):1600-1608. doi: 10.1111/1756-185X.13331. Epub
[PubMed PMID: 30146740]
[72]
Ahmad K, Nathan SD. Novel management strategies for idiopathic pulmonary fibrosis. Expert review of respiratory medicine. 2018 Oct:12(10):831-842. doi: 10.1080/17476348.2018.1513332. Epub 2018 Aug 30
[PubMed PMID: 30136607]