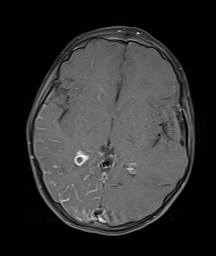
[1]
Comi AM. Sturge-Weber syndrome. Handbook of clinical neurology. 2015:132():157-68. doi: 10.1016/B978-0-444-62702-5.00011-1. Epub
[PubMed PMID: 26564078]
[2]
Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. The New England journal of medicine. 2013 May 23:368(21):1971-9. doi: 10.1056/NEJMoa1213507. Epub 2013 May 8
[PubMed PMID: 23656586]
[3]
Pinto A, Sahin M, Pearl PL. Epileptogenesis in neurocutaneous disorders with focus in Sturge Weber syndrome. F1000Research. 2016:5():. pii: F1000 Faculty Rev-370. doi: 10.12688/f1000research.7605.1. Epub 2016 Mar 18
[PubMed PMID: 27019697]
[4]
Enjolras O, Riche MC, Merland JJ. Facial port-wine stains and Sturge-Weber syndrome. Pediatrics. 1985 Jul:76(1):48-51
[PubMed PMID: 4011357]
[5]
Maraña Pérez AI, Ruiz-Falcó Rojas ML, Puertas Martín V, Domínguez Carral J, Carreras Sáez I, Duat Rodríguez A, Sánchez González V. Analysis of Sturge-Weber syndrome: A retrospective study of multiple associated variables. Neurologia (Barcelona, Spain). 2017 Jul-Aug:32(6):363-370. doi: 10.1016/j.nrl.2015.12.012. Epub 2016 Mar 8
[PubMed PMID: 26964511]
Level 2 (mid-level) evidence
[6]
Comi AM. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphatic research and biology. 2007:5(4):257-64. doi: 10.1089/lrb.2007.1016. Epub
[PubMed PMID: 18370916]
[7]
Chugani HT, Mazziotta JC, Phelps ME. Sturge-Weber syndrome: a study of cerebral glucose utilization with positron emission tomography. The Journal of pediatrics. 1989 Feb:114(2):244-53
[PubMed PMID: 2783735]
[8]
Arzimanoglou AA, Andermann F, Aicardi J, Sainte-Rose C, Beaulieu MA, Villemure JG, Olivier A, Rasmussen T. Sturge-Weber syndrome: indications and results of surgery in 20 patients. Neurology. 2000 Nov 28:55(10):1472-9
[PubMed PMID: 11094100]
[9]
Bay MJ, Kossoff EH, Lehmann CU, Zabel TA, Comi AM. Survey of aspirin use in Sturge-Weber syndrome. Journal of child neurology. 2011 Jun:26(6):692-702. doi: 10.1177/0883073810388646. Epub 2011 Mar 22
[PubMed PMID: 21427442]
Level 3 (low-level) evidence
[10]
Thavikulwat AT, Edward DP, AlDarrab A, Vajaranant TS. Pathophysiology and management of glaucoma associated with phakomatoses. Journal of neuroscience research. 2019 Jan:97(1):57-69. doi: 10.1002/jnr.24241. Epub 2018 Apr 1
[PubMed PMID: 29607552]
[11]
Higueros E, Roe E, Granell E, Baselga E. Sturge-Weber Syndrome: A Review. Actas dermo-sifiliograficas. 2017 Jun:108(5):407-417. doi: 10.1016/j.ad.2016.09.022. Epub 2017 Jan 23
[PubMed PMID: 28126187]