Continuing Education Activity
Leukocytoclastic vasculitis is a cutaneous, small-vessel vasculitis of the dermal capillaries and venules. This condition can be idiopathic or can be associated with infections, neoplasms, autoimmune disorders, and drugs. Key clinical features of leukocytoclastic vasculitis include palpable purpura on the lower extremity, small vessel involvement, and, in about 30 percent of individuals, extracutaneous involvement. Most cases of idiopathic cutaneous, small vessel vasculitis are self-limited with 90 percent of cases resolving in weeks to months of onset. Otherwise, treatment depends on the severity of disease and can range from an oral corticosteroid taper to various steroid-sparing immunosuppressive agents. This activity describes the evaluation and management of leukocytoclastic vasculitis and highlights the role of the interprofessional team in the care of affected patients.
Objectives:
- Describe the etiology of leukocytoclastic vasculitis.
- Review the typical presentation of leukocytoclastic vasculitis.
- Outline the management options available for leukocytoclastic vasculitis.
- Explain interprofessional team strategies for improving care coordination and communication to advance the treatment of leukocytoclastic vasculitis and improve outcomes.
Introduction
Vasculitis refers to inflammation of the blood vessels leading to tissue destruction with or without organ damage. Vasculitis is classified as small vessel, medium vessel or large vessel vasculitis[1] and maybe either idiopathic or associated with an underlying pathology/disease. Small vessel vasculitis can be seen secondary to systemic vasculitides such as Anti-neutrophil Cytoplasmic Antibody (ANCA) associated vasculitis (Microscopic polyangiitis, Granulomatosis with polyangiitis or Eosinophilic granulomatosis with polyangiitis), Behçet’s disease, and Cogan’s syndrome. Immune complex-mediated small vessel vasculitis can be seen in rheumatoid arthritis, systemic lupus erythematosus, Sjogren syndrome, Henoch-Schönlein purpura, cryoglobulinemic vasculitis, Hypocomplementemic urticarial vasculitis, Erythema elevatum diutinum, and cutaneous leukocytoclastic angiitis, formerly known as hypersensitivity vasculitis. Other causes of small vessel vasculitis or leukocytoclastic vasculitis include drug-induced vasculitis, paraneoplastic vasculitis, and infection associated vasculitis (hepatitis B, hepatitis C, syphilis).
Leukocytoclastic vasculitis is a small vessel vasculitis characterized histopathologically by immune complex-mediated vasculitis of the dermal capillaries and venules. Cutaneous leukocytoclastic vasculitis is usually confined to skin with rare extracutaneous manifestations in less than 30% of the cases. Key clinical features of cutaneous leukocytoclastic angiitis include palpable purpura, lower extremity location, small vessel involvement.[2][3][4] If leukocytoclastic vasculitis is suspected, a punch biopsy should be performed with direct immunofluorescence studies. If no systemic symptoms are present, laboratory testing including C-reactive protein, complete blood count (CBC), basic metabolic panel, liver function tests, and urinalysis should be done as well. If there is a concern for systemic involvement, a more extensive workup needs to be performed. Most cases of idiopathic cutaneous small-vessel vasculitis cases are self-limited with 90% resolving in weeks to months of onset. In persistent vasculitis, treatment depends on the severity of disease and can range from oral corticosteroids to various steroid-sparing agents.[5][6]
Etiology
Leukocytoclastic vasculitis is idiopathic up to 50% of the cases. Infections and drugs are the most common triggers for secondary leukocytoclastic vasculitis.[7] It can be seen secondary to underlying systemic autoimmune diseases, chronic infections, and malignancies as well. Post-infectious leukocytoclastic vasculitis is most commonly seen after streptococcal upper respiratory tract infection. Other infectious triggers include, but are not limited to, Mycobacterium, Staphylococcus aureus, Chlamydia, Neisseria, and HIV. Chronic infections with hepatitis B, hepatitis C, and syphilis can be associated with leukocytoclastic vasculitis as well. Several drugs have been associated with leukocytoclastic vasculitis. The onset is typically 1 to 3 weeks after drug initiation. These include, but are not limited to, beta-lactams, erythromycin, clindamycin, vancomycin, sulfonamides, furosemide, allopurinol, NSAIDs, amiodarone, gold, thiazides, phenytoin, beta-blockers, TNF-alpha inhibitors, selective serotonin reuptake inhibitors, metformin, warfarin, valproic acid, among many others. Neoplastic or hematologic triggers of leukocytoclastic vasculitis include, but are not limited to lymphomas, leukemias, visceral tumors such as intestinal adenocarcinoma and lung cancer. Systemic diseases including connective tissue diseases (systemic lupus erythematosus, Sjogren syndrome), inflammatory bowel disease, Behcet disease, and rheumatoid arthritis cryoglobulinemic vasculitis, Henoch-Schönlein purpura (HSP), Hypocomplementemic urticarial vasculitis, and Erythema elevatum diutinum are also associated with leukocytoclastic vasculitis.[8]
Epidemiology
The annual incidence of biopsy-proven leukocytoclastic vasculitis is approximately 45 per million individuals. The epidemiology of leukocytoclastic vasculitis varies with the underlying etiology. Leukocytoclastic vasculitis occurs in all ages and both genders; however, it typically presents in adults. However, HSP is usually seen in children and young adults with an incidence of up to 270 cases per million children per year with a slight male predominance.
Pathophysiology
The pathogenesis of leukocytoclastic vasculitis involves immune complex deposition in small vessel walls in addition to activation of the complement system. Neutrophils are recruited, and injury to vessel walls ensues with secondary exudation of erythrocytes, fibrin, and serum. Fibrinoid necrosis of small vessel walls will be noted secondary to lysosomal enzymes such as collagenases and elastases as well as reactive oxygen species. Lymphokines aid in the evolution of the clinical findings as well. IL-1, IL-6, IL-8, and tumor necrosis factor are increased in circulation. Turbulence and increased venous pressure present in the lower extremities can elucidate why leukocytoclastic vasculitis commonly tends to occur on the legs.[9][10][11]
Histopathology
The histopathologic features of leukocytoclastic vasculitis will vary based on the time frame of the vasculitis. Fresh lesions biopsied within the 1st 18-24 hours of onset, from a nonulcerated site have the highest yield with most diagnostic findings. On light microscopy, findings of vessel wall destruction by the infiltration of inflammatory cells within and around the vessel wall can be seen. Classically leukocytoclastic vasculitis demonstrates neutrophil infiltration in the small vessel walls. The neutrophils undergo degeneration, known as leukocytoclasis with nuclear dust (karyorrhexis). Fibrinoid necrosis of the vessel walls can be apparent around the vasculature. Extravasation of red blood cells can be present in the dermis as well. In drug-related cases, eosinophils are often noted in the dermis. Older lesions especially those more than 48 hours old can demonstrate lymphocytic infiltration.
Direct immunofluorescence is strongly recommended in cases of new-onset leukocytoclastic vasculitis. While a negative result carries a low yield, a positive result can sometimes be diagnostic of an underlying disease and can provide insight into the underlying disease pathophysiology. Further, light microscopy and histopathological features of leukocytoclastic vasculitis may not be sufficient in differentiating pauci-immune vasculitis from immune complex-mediated vasculitis. Immunofluorescence shall be performed with fluorescein-labeled antibodies against IgG, IgM, IgA, and C3. Strong IgA deposition without other antibody deposition is indicative of HSP. Leukocytoclastic vasculitis associated with underlying systemic lupus erythematosus can show diffusely positive immunofluorescence in addition to increased dermal mucin.
History and Physical
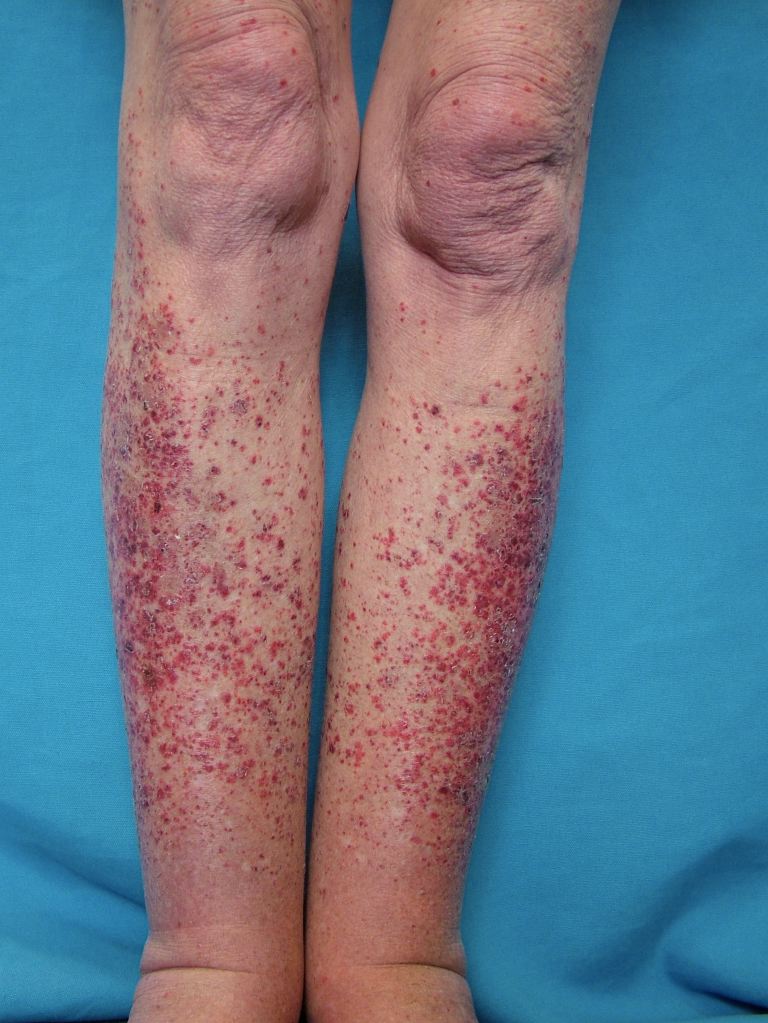
The cutaneous manifestations of leukocytoclastic vasculitis usually appear about 1-3 weeks after the triggering event. Leukocytoclastic vasculitis presents as erythematous macules with palpable purpura bilaterally on dependent areas of the body like the lower extremities and buttocks. Unilateral presentations and localized lesions are rare. Hemorrhagic vesicles and bulla, pustules, nodules, crusted ulcers, or livedo reticularis may also be present on physical exam. The lesions can range in size from 1 mm to 1 cm in diameter. The lesions can appear at once in crops, or various crops may cycle and produce lesions with different stages of evolution. Koebnerization, the appearance of lesions at areas of trauma, is uncommon with this vasculitis. In fact, reverse koebnerization has been described with the disappearance of the lesions with pressure bandage application after the biopsy. The lesions are asymptomatic but may itch, burn, or sting.
Extracutaneous manifestations with leukocytoclastic vasculitis are less common. Systemic symptoms noted with leukocytoclastic vasculitis may include low-grade fevers, malaise, weight loss, myalgias, and arthralgias. These findings have been noted in approximately 30% of affected patients, with arthralgias compromising the most common manifestation. Other less common manifestations include renal, gastrointestinal, pulmonary, or neurological symptoms, and these entities may be better classified as microscopic polyangiitis/polyarteritis. Other features of the underlying disease associated with leukocytoclastic vasculitis can give a clue to the underlying diagnosis. Leukocytoclastic vasculitis in children along with symptoms of inflammatory arthritis, abdominal pain, edema and hematuria/renal disease is characteristic of Henoch-Schoenlein purpura. Leukocytoclastic vasculitis is unlikely to be the presenting complaint in rheumatoid arthritis and Sjogren syndrome and usually, these patients already have a well-established diagnosis for several years before they developed leukocytoclastic vasculitis. In systemic lupus erythematosus, leukocytoclastic vasculitis is less likely to be the presenting initial complaint but it can be seen as the initial complaint. These patients will likely have other associated features of systemic lupus erythematosus as well.
Evaluation
Skin biopsy with direct immunofluorescence is the cornerstone for the diagnosis of leukocytoclastic vasculitis. Workup for an underlying disease should be undertaken based on clinical suspicion. Complete blood counts, liver function test and renal function tests along with urinalysis are recommended at least once at baseline. A more extensive workup shall be pursued if systemic involvement is a concern including a viral hepatitis panel, HIV testing, ASO titer (if a streptococcal infection is the suspected trigger), antinuclear antibody and more specific antibodies for systemic lupus erythematosus and Sjogren syndrome, rheumatoid factor, anti-CCP antibody, serum complements, serum protein electrophoresis and cryoglobulins.
Treatment / Management
Most cases of idiopathic cutaneous leukocytoclastic vasculitis are mild and resolve with supportive measures such as leg elevation, rest, compression stockings, and antihistamines. In more chronic or resistant cases, a 4-6 week tapering dose of corticosteroids can be used. Rarely, immunosuppressive steroid-sparing agents such as methotrexate, azathioprine, mycophenolate mofetil, dapsone, cyclophosphamide, and intravenous immunoglobulin may be needed.
If an offending drug has been identified, withdrawal of the drug is crucial in the resolution of the vasculitis. In cases of leukocytoclastic vasculitis associated with an infection, treatment of the infection usually results in the resolution of the vasculitis. Similarly, malignancy-associated leukocytoclastic vasculitis improves with treatment of the malignancy. When leukocytoclastic vasculitis is associated with an underlying connective tissue disease or an autoimmune disease such as systemic lupus erythematosus or rheumatoid arthritis, better control of the underlying disease by an escalation of the immunosuppressive therapy may be needed to treat the leukocytoclastic vasculitis and prevent relapses. Mild cases of HSP usually do not require corticosteroids or immunosuppressive agents, and skin rash and arthralgia usually respond to NSAIDs. High-dose corticosteroids and/or immunosuppressive agents may be indicated in cases of severe renal involvement or systemic involvement.
Differential Diagnosis
The differential diagnosis includes:
- Thrombocytopenic purpura
- Benign pigmented purpura
- Schamberg disease
- This is an idiopathic phenomenon, which results from erythrocyte extravasation into the dermis in the lower extremities due to capillary fragility or leaky blood vessels, venous stasis or exercise. Hyperpigmentation in the lower extremities is frequently present.
Skin biopsy can easily differentiate benign pigmented purpura from leukocytoclastic vasculitis given the lack of features of vasculitis such as fibrinoid necrosis and vessel wall disruption by the inflammatory infiltrates.[12]
Prognosis
The mortality rate of leukocytoclastic vasculitis is low (about 2%), and related to systemic involvement versus strictly cutaneous involvement. Approximately 90% of patients experience spontaneous resolution of their skin lesions within weeks to months, with the remaining 10% continuing to chronic disease averaging 2 to 4 years. Arthralgias without fever may portend chronicity.
Complications
Rare complications of cutaneous leukocytoclastic vasculitis include skin ulcerations which may get secondarily infected. While wound care is crucial in the management of these skin ulcers, they usually do not resolve unless immunosuppressive therapy such as corticosteroids is used to treat the underlying vasculitic insult.
Deterrence and Patient Education
Early identification, especially in drug-induced leukocytoclastic vasculitis, is crucial and patients should be educated about this potential adverse effect of the medications before initiating the medication. Patients with idiopathic cutaneous leukocytoclastic vasculitis usually carry a favorable prognosis, however, if there is a secondary etiology, close follow-up is needed and patients should be educated about the importance of compliance and regular monitoring. Further, patients should be educated about the course of the leukocytoclastic vasculitis as it may take several weeks to months for it to resolve.
Enhancing Healthcare Team Outcomes
The involvement of an interprofessional team including physicians, nurse practitioners, nurses, and pharmacists is needed to manage leukocytoclastic vasculitis. Pharmacists can assist in identifying potential drug triggers and removing them. The involvement of the nursing team is crucial in maintaining close follow-up and monitoring of the patient. The involvement of specialists including rheumatologists may be needed in some cases especially when secondary etiologies are suspected. Finally, patient education and reassurance about the benign course of the idiopathic cutaneous leukocytoclastic vasculitis as in most cases can help prevent undue anxiety in the patients. Close communication among the team members can significantly enhance patient-centered care. [Level V]