Continuing Education Activity
Dermatomyositis is a rare condition that causes muscle inflammation. It presents with symmetric proximal muscle weakness, skin rash, and extramacular manifestations, such as esophageal dysfunction and interstitial lung disease. Dermatomyositis is strongly associated with malignancy, especially in adults. This activity outlines the evaluation and management of dermatomyositis and highlights the role of the interprofessional team in improving care for patients with this condition.
Objectives:
- Describe the histopathology of dermatomyositis.
- Identify the typical history and physical exam findings associated with dermatomyositis.
- Review the complications of dermatomyositis, such as malignancy and systemic involvement.
- Outline the importance of improving care coordination among the interprofessional team to improve outcomes for patients affected by dermatomyositis.
Introduction
Dermatomyositis is a rare acquired immune-mediated muscle disease characterized by muscle weakness and skin rash. It is classified as one of the idiopathic inflammatory myopathies (IIM). Although all idiopathic inflammatory myopathies share the common presentation of muscle weakness, they differ clinically in terms of muscle groups involved and histopathological findings.[1][2] Dermatomyositis presents with characteristic skin findings and symmetric proximal skeletal muscle weakness. Also, it can affect other organ systems such as the pulmonary, cardiovascular, and gastrointestinal systems. A significant proportion of patients with dermatomyositis have an underlying malignancy, which can alter the prognosis of the condition. Although a majority of cases have muscular and cutaneous manifestations, other variants of the condition exist. Clinically amyopathic dermatomyositis (CADM) is a condition in which patients have the characteristic cutaneous findings of dermatomyositis, but do not have muscle weakness. Clinically amyopathic dermatomyositis is further classified as hypomyopathic or amyopathic dermatomyositis. Patients with hypomyopathic dermatomyositis lack muscle weakness clinically. However, there is evidence of myositis based on laboratory investigations, electromyography, or muscle biopsy. In contrast, patients with amyopathic dermatomyositis lack both clinical and laboratory evidence of muscle involvement.[3]
Etiology
Although the cause of dermatomyositis is unknown, several genetic, immunologic, and environmental factors are implicated in this condition.
Genetic Factors
Multiple studies have indicated that patients with particular human leukocyte antigen (HLA) types are at higher risk of dermatomyositis. High-risk haplotypes include HLA-A*68 in North American Whites[4], HLA-DRB1*0301 in African Americans,[5] HLA-DQA1*0104 and HLA-DRB1*07 in Han Chinese,[6] DQA1*05 and DQB1*02 in people from the UK. Also, DRB1*03-DQA1*05-DQB1*02 haplotype is strongly associated with the development of interstitial lung disease in dermatomyositis.[7]
Immunologic Factors
Although autoantibodies are detected in patients with dermatomyositis, it is unclear whether they play a role in pathogenesis.
Environmental Factors
Infections
Viruses such as Coxsackie B virus, enterovirus, and parvovirus have been suspected of acting as triggers of dermatomyositis. There are multiple theories about the mechanisms of virus-induced autoimmunity. These include alteration of cellular proteins, breakdown of self-tolerance, an unmasking of previously hidden epitopes, autoantibody induced B cell activation, and molecular mimicry.[8]
Drugs
Several drugs can trigger dermatomyositis. These include antineoplastic drugs (hydroxyurea, cyclophosphamide), anti-infectious agents (penicillin, sulfonamides, isoniazid), non-steroidal anti-inflammatory drugs (diclofenac, phenylbutazone), D-penicillamine, statins, and certain vaccines.[9]
Radiation
Dermatomyositis has been observed to occur more frequently among women exposed to high-intensity ultraviolet radiation.[10]
Epidemiology
Dermatomyositis is a rare condition. A retrospective study conducted between 1967 and 2007 in Olmsted county, Minnesota, estimated an incidence rate of 9.63 per 1,000,000 people. The same study also found that 21% of all cases were of the amyopathic subtype. Dermatomyositis commonly affects persons between the ages of 40 and 50 with a mean age at diagnosis of 44.0 ± 18.3 years. The condition is more common in women than in men, with incidence rates being 3.98 and 4.68 per 1,000,000, respectively.[11][12]
In Europe, a higher prevalence of dermatomyositis has been noted in Southern Europe compared to Northern Europe.[13] A study conducted in Quebec showed a higher prevalence of dermatomyositis in urban areas. A cohort study conducted in Pennsylvania also showed clusters of clinically amyopathic dermatomyositis (CADM) in regions with high airborne pollution. These studies point towards the possibility of environmental factors acting as triggers for the condition.[14][15]
Pathophysiology
Dermatomyositis is thought to be the result of a humoral mediated attack directed against the muscle capillaries and the endothelium of arterioles. The initiating event is the activation of completer factor-3 (C3), which forms C3b and C4b. This is followed by the formation of the neoantigen C3bNEO and the C5b-C9 membrane attack complex (MAC). The membrane attack complex deposits on vascular walls and causes inflammation. Hypoxic injury to the muscle fibers ensues, leading to atrophy of muscle fibers, particularly the fibers at the periphery that are the most remote and away from the vascular supply. Over time, the capillary density reduces, and muscle fibers start to undergo necrosis and degeneration.
Histopathology
Muscle Biopsy
Muscle biopsy often shows the following findings, which can be diagnostic:
- Perivascular and perimysial inflammatory infiltrate: The infiltrate in dermatomyositis is concentrated around the perivascular and interfascicular regions and consists of B cells, CD4+ T helper cells, macrophages, and plasmacytoid dendritic cells. In contrast to polymyositis, CD8+ T cells and NK cells are rarely present.
- Perifascicular atrophy: Atrophy of muscle fibers, especially around the periphery of fascicles, is a hallmark histopathological feature of dermatomyositis. Degenerating and regenerating muscle fibers may be observed in the perifascicular region.
- Microangiopathy: Injury to intramuscular blood vessels takes the form of immunoglobulin and complement (C5b-C9 membrane attack complex) deposits on endomysial capillaries. A reduced capillary density and endothelial hyperplasia may be observed.[16][17]
Skin Biopsy
Skin biopsy findings in dermatomyositis are similar to those found in systemic lupus erythematosus. Typical findings include vacuolar changes of the basal layer, increased lymphocytic infiltrate, and increased mucin deposition in the dermis.[18]
History and Physical
A comprehensive history and physical exam should be conducted in suspected cases of dermatomyositis, keeping in mind the following objectives:
- Identify the typical muscular and cutaneous signs and symptoms of dermatomyositis
- Exclude other causes of muscle weakness e.g., inherited, infectious or endocrine myopathy
- Conduct a detailed review of systems to determine if other organ systems are involved (respiratory, cardiac, esophageal)
- Evaluate for signs and symptoms of a possible underlying malignancy and perform age-appropriate cancer screening when indicated
Muscle weakness and skin findings comprise the main presenting symptoms in dermatomyositis. The onset of the disease may be insidious or acute with a waxing and a waning course.
Muscular
Muscle weakness is the most common presenting symptom in dermatomyositis. The weakness usually has a subacute onset with the development of gradually progressive symmetric proximal muscle weakness. Patients may report difficulty in carrying out activities such as climbing stairs, getting up from a seated position, lifting objects, combing hair, and raising their head from a pillow. Distal muscle weakness, muscle pain, and stiffness are uncommon in dermatomyositis. In severe cases, dysphagia, or dysphonia may be present. The examination may reveal reduced muscle strength of proximal muscles, such as the deltoids, hip flexors, and neck flexors. Usually, muscle tenderness is mild, and distal muscle strength is preserved. Depressed deep tendon reflexes and muscle atrophy are not seen unless the disease is severe and long-standing.
Cutaneous
Skin changes may precede or may coincide with the onset of muscular symptoms. Patients can present with several types of skin rashes, photosensitivity, changes in pigmentation, and pruritis. Dermatomyositis can also cause nail changes and alopecia.
Pathognomonic findings of dermatomyositis include the following:
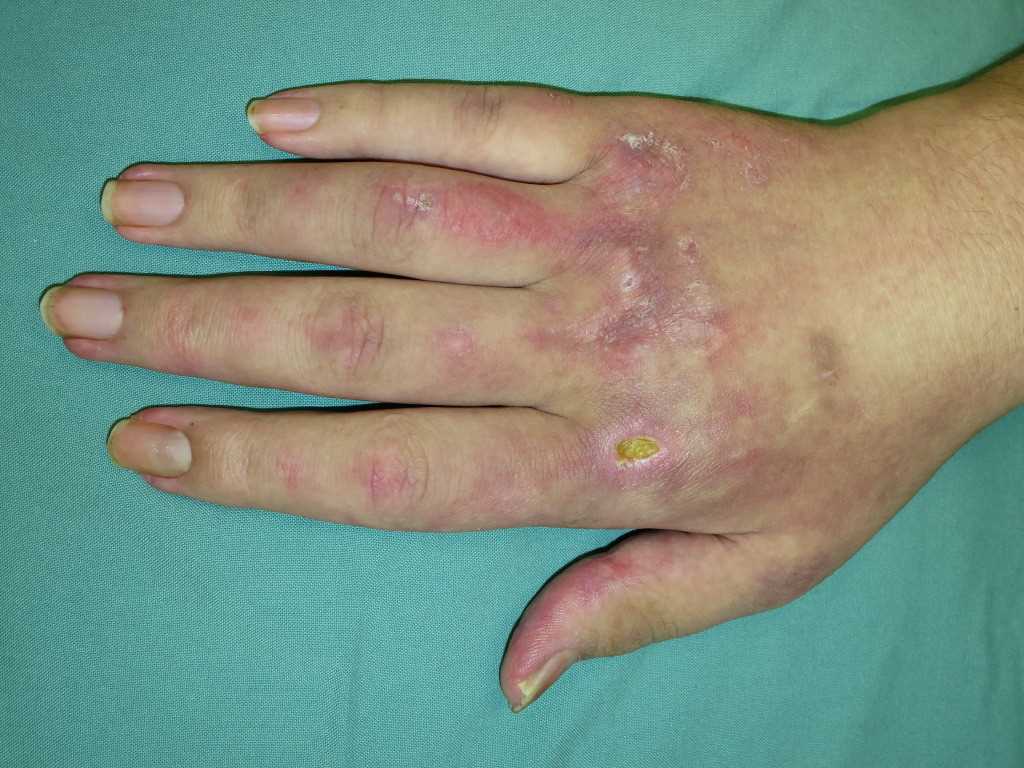
- Gottron papules: dorsal metacarpophalangeal and interphalangeal joints may show the presence of overlying erythematous or violaceous papules with or without scaling or ulceration.
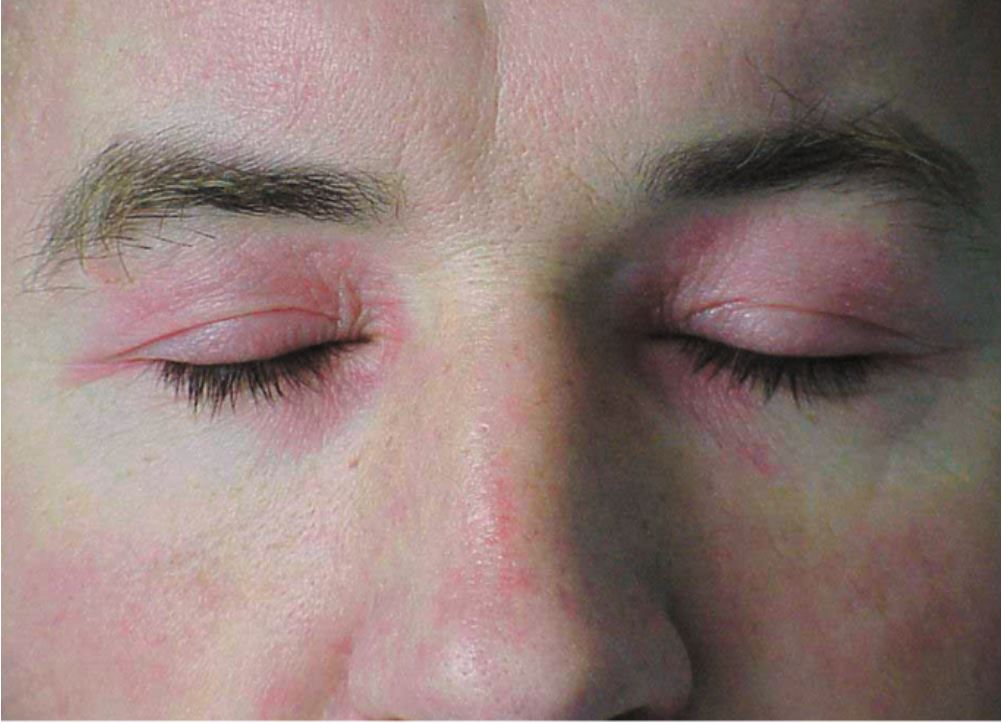
- Heliotrope rash: This is a characteristic skin finding of dermatomyositis and presents with a violaceous, or an erythematous rash affecting the upper eyelids with or without periorbital edema. This finding may not be apparent in patients with dark skin patients.
Other skin findings that may help differentiate dermatomyositis from other conditions include the following:
- Gottron sign: erythematous macules or patches over the elbows or knees
- Facial erythema: erythema over the cheeks and nasal bridge involving the nasolabial folds. The rash may extend up to the forehead and laterally up to the ears.
- Shawl sign: erythema over the posterior aspect of the neck, upper back, and shoulders at times, extending to the upper arms.
- V sign: ill-defined erythematous macules involving the anterior aspect of the neck and the upper chest.
- Poikiloderma: atrophic skin with changes in pigmentation and telangiectasia in photo-exposed or non-exposed areas.
- Holster sign: poikiloderma involving the lateral aspects of the thighs.
- Periungual involvement: telangiectasias and cuticular overgrowth
- Mechanic's hands: hyperkeratotic, cracked horizontal lines on the palmar and lateral aspects of the fingers.
- Scalp involvement: diffuse poikiloderma, with scaling and pruritis.
- Calcinosis cutis: calcium deposits in the skin
Joints
Dermatomyositis can cause non-erosive polyarthritis or arthralgia of the small joints of the hands. Patients may present with joint pain or swelling.
Respiratory
Patients may present with exertional dyspnea, exercise intolerance, and non-productive cough due to underlying interstitial lung disease (ILD). Auscultation of the chest may reveal the presence of bilateral dry crackles. Reduced chest movement may be seen due to respiratory muscle weakness.
Esophageal
Patients may report difficulty swallowing solids and liquids due to the weakness of the muscles of the oropharynx and upper esophagus. They may also have symptoms of gastroesophageal reflux.
Other findings
Other findings that may present in dermatomyositis include Raynaud's phenomenon, gastrointestinal ulcers, and cardiac symptoms. Systemic symptoms such as fever, malaise, and weight loss may be present, which may indicate an occult malignancy. The following factors may predict malignancy; male gender, older age at onset, the presence of dysphagia, and the absence of interstitial lung disease.
Patients should be inquired about the intake of any drugs and their family history. Patients should also undergo age-appropriate cancer screening exams such as pelvic exams, breast exams, testicular exams, and rectal exams [19].
Evaluation
Lab Investigations
Muscle Enzymes
Initial testing in suspected cases of dermatomyositis should include muscle enzymes, such as creatine kinase (CK), aldolase, lactate dehydrogenase (LDH), aspartate aminotransferase (AST) and alanine aminotransferase (ALT). Testing for muscle enzymes helps to guide further diagnostic studies and to assess response to therapy. In some cases, the elevation of muscle enzymes occurs prior to the appearance of muscle weakness.
Autoantibodies
Antinuclear antibodies (ANA) are present in a majority of patients with dermatomyositis but do not help to make a diagnosis. Instead, testing should focus on detecting myositis specific autoantibodies (MSA), which are present in approximately 30% of dermatomyositis and polymyositis patients. Testing for myositis-specific antibodies offers valuable information for determining the prognosis and can help to predict the pattern of organ involvement. Aminoacyl-transfer (t) ribonucleic acid synthetase (also known as antisynthetase antibody) is the most common myositis specific autoantibody associated with dermatomyositis. Anti-Jo is the most common antisynthetase antibody found in dermatomyositis. The following autoantibodies are associated with specific complications and findings:
- Anti-Jo: antisynthetase syndrome consisting of interstitial lung disease, mechanic's hands, Raynaud phenomenon, sclerodactyly, and arthritis.
- Anti-Mi2 (directed against-helicase): Acute onset disease, V-neck sign, and shawl rash
- Anti- SRP (directed against signal recognition particle): severe myositis, resistant to treatment
- Anti- MDA5 (melanoma differentiation-associated gene 5): severe cutaneous involvement, amyopathic dermatomyositis, and rapidly progressive ILD
- Anti- TIF-1 gamma (transcription intermediary factor) /Anti-p155/140: malignancy
- Anti-SAE (ubiquitin-like modifier activating enzyme): dysphagia, skin disease preceding myositis
- Anti-NXP2 (nuclear matrix protein 2): calcinosis cutis[20]
Electromyography (EMG)
Electromyography helps to identify which groups of muscles are most affected and provides guidance about which muscles to biopsy. It also helps to distinguish dermatomyositis from neuropathic conditions. However, the electromyographic findings are not specific and may be absent in 11% of patients.[21] Findings suggestive of dermatomyositis include the following:
- Increased insertional activity
- Spontaneous fibrillations
- Positive sharp waves
- Complex repetitive discharges
- Early recruitment
- Low-amplitude, short polyphasic motor unit potentials
Radiology
Chest radiography: Every patient with dermatomyositis should undergo chest radiography to screen for interstitial lung disease. If the patient has respiratory symptoms or abnormal chest X-ray findings, further testing with high-resolution computer tomography (HRCT) of the chest. In addition, pulmonary function tests should be performed. Findings on HRCT suggestive of interstitial lung disease include nodules, fibrosis, linear opacities, honey-combing, or consolidation.
Magnetic resonance imaging (MRI): Magnetic resonance imaging of skeletal muscles is a non-invasive and sensitive test to evaluate myositis. Typical findings include muscle edema, areas of inflammation; that appear hyperintense on T2-weighted images; and fat suppression.[22]
Barium swallow: may be done if esophageal dysfunction is present.
Histopathology
Muscle biopsy is the most accurate test to confirm the diagnosis of dermatomyositis and to exclude other causes of muscle weakness or skin rash. However, choosing the right muscle for a biopsy is crucial to prevent a missing diagnosis. Muscle biopsy should be obtained on weak muscles as identified by physical exam or contralateral to the abnormal muscles, as identified by electromyography. A muscle biopsy should be obtained from patients with suspected dermatomyositis but who lack the characteristic skin findings. Similarly, patients who have the characteristic skin manifestations of dermatomyositis but lack muscle weakness should undergo a skin biopsy procedure.[18]
Other Investigations
Other baseline lab investigations include a complete blood count with differential, creatinine, liver function tests, and inflammatory markers such as erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). ESR in dermatomyositis is usually normal, or may only be mildly elevated. Serum Thyroid-stimulating hormone (TSH) may be ordered to exclude hypothyroidism. Electrocardiography (ECG) may be ordered to look for conduction abnormalities that might be subclinical. Pulmonary function tests may be conducted to assess the severity of pulmonary involvement. Patients with interstitial lung disease show a restrictive defect on pulmonary function tests with reduced forced vital capacity (FVC), reduced total lung capacity (TLC), and diminished diffusing capacity. Patients with respiratory muscle weakness will also show a restrictive pattern, but they may have normal diffusing capacity.
Investigations for Malignancy
Patients with dermatomyositis are at the highest risk of having an underlying malignancy during the first five years of diagnosis. Therefore, all patients should undergo age and sex appropriate cancer screening at diagnosis regardless of whether or not, and they have specific symptoms. Initial testing for malignancy may include colonoscopy (or fecal occult blood test), urine analysis, mammography, and pap smears. Women at high risk of ovarian cancer should be screened, with serial measurements of CA-125 and transvaginal ultrasound. There is no consensus on the frequency and how extensively patients with dermatomyositis should undergo cancer screening. Earlier studies recommend against extensive evaluation for malignancy apart from history, physical exam, and basic lab tests.[23] However, more recent evidence indicates that blind testing with computer tomography of the chest, abdomen, and pelvis in asymptomatic patients may be valuable in detecting occult malignancies.[24]
Classification Criteria
Historically, several classification criteria have been used to identify idiopathic inflammatory myopathies, including dermatomyositis. These classifications systems include the Bohan and Peter classification (1975), Targoff criteria (1997), and the Dalakas and Hohlfeld's criteria (2003). However, most of these classification systems suffer from limitations ranging from a vague description of skin findings, unclear exclusion criteria, failure to incorporate the presence of autoantibodies as a criterion, and failure to include all subgroups of idiopathic inflammatory myopathies.[25][26]
In 2017, the European League Against Rheumatism and the American College of Rheumatology (EULAR/ACR) released a set of criteria to help identify idiopathic inflammatory myopathies and its major subgroups. This new classification system overcomes many of the limitations of the previous classification systems. The EULAR/ACR criteria for idiopathic inflammatory myopathies have been found to have a sensitivity of 87% (93% with muscle biopsy) and specificity of 82% (88% with muscle biopsy). According to this classification, a combination of clinical, lab, and muscle biopsy criteria are used to calculate the probability of having idiopathic inflammatory myopathies.
Clinical criteria include the age at onset of symptoms, the pattern of muscle weakness (proximal, progressive, symmetric muscle weakness involving upper limbs/lower limbs, selective involvement of neck flexors), presence of skin manifestations (heliotrope rash, Gottron papules, Gottron sign) and other findings (esophageal dysmotility, dysphagia). Lab criteria include the presence of anti–Jo-1 (anti–histidyl–transfer RNA synthetase) and elevation of muscle enzymes (creatine kinase, lactate dehydrogenase, aspartate transaminase, alanine aminotransferase).
A probability of 55% or more is considered the threshold for the diagnosis of idiopathic inflammatory myopathy. Patients are further classified as having "possible" "probable" or "definite" idiopathic inflammatory myopathy if the probabilities are 50% to less than 55%, 55% to 75% (including muscle biopsy findings), and greater than 90%, respectively. Once the minimum criteria for the diagnosis of idiopathic inflammatory myopathy are met, subtypes such as dermatomyositis can be identified using a sub-classification tree.[27]
Treatment / Management
The goals of managing dermatomyositis are focused on treating muscle weakness, skin disease, and addressing any other underlying complications.
The first-line treatment of muscle disease in dermatomyositis is systemic glucocorticoids with or without immunosuppressants. Although there is no standard systemic steroid regimen specified for dermatomyositis, the general principles of therapy are the same. Initially, prednisolone is given at high doses for the first few months until the muscle enzyme levels decline, and muscle strength improves. During this time, patients should be regularly evaluated for an adequate response, keeping in mind that it takes approximately six weeks for muscle enzymes to normalize. Also, it may require as long as three months for muscle weakness to improve. Once an adequate response occurs, the administration of systemic steroids is gradually tapered off over time. The total duration of therapy with systemic steroids usually spans between nine and twelve months. It is important to note that administering high dose glucocorticoids for more than six weeks may lead to glucocorticoid myopathy.
A failure to respond to initial treatment with systemic steroids should prompt the physician to exclude other causes of myopathy. These may include hypothyroidism, inclusion body myositis, glucocorticoid myopathy, or underlying malignancy. Once alternate diagnoses are ruled out, steroid-sparing immunosuppressants can be added.
Immuno-suppressants help to alleviate the adverse effects of long-term systemic steroids. Such adverse effects could include osteoporosis, increased susceptibility to infections, cushingoid features, and secondary diabetes. Immunosuppressant administration can be initiated along with steroids in cases of severe myopathy, chronic medical conditions (e.g., diabetes), and profound extra-muscular complications (e.g., interstitial lung disease, esophageal dysfunction). First-line agents include azathioprine and methotrexate. The choice of immunosuppressant depends on many factors, including dosing frequency, systemic involvement, adverse effects, and alcohol use. Administration of an immuno-suppressant such as azathioprine is preferred in patients with liver involvement, interstitial lung disease, and those who are unable to abstain from alcohol. On the other hand, methotrexate has the advantage of once-a-week dosing.
Patients who do not respond satisfactorily to therapy with steroids and azathioprine or methotrexate are considered resistant. Treatment options for resistant cases include rituximab, mycophenolate mofetil, calcineurin inhibitors, intravenous immunoglobulin (IVIG), and cyclophosphamide. Rituximab is an anti-CD 20 agent and is the recommended first-line agent in resistant cases. If this fails, intravenous immunoglobulin or a combination of azathioprine and methotrexate can be used as second-line therapy. Mycophenolate mofetil and tacrolimus are useful in refractory cases, especially if there is a concomitant interstitial lung disease. Cyclophosphamide is preferred in cases of rapidly progressive interstitial lung disease.
It is essential to monitor patients for any adverse effects of immunosuppressants. For example, patients on methotrexate should be monitored for stomatitis, hepatotoxicity, and leucopenia. Cotreatment with folic acid or leucovorin can help to minimize these adverse effects. Azathioprine can cause a flu-like reaction, which may require discontinuation of therapy. It also causes myelosuppression and pancreatitis. Cyclophosphamide increases the risk of malignancy and should be avoided unless multiple drug therapies have failed.
Skin disease in dermatomyositis is managed with general measures, physiotherapy, and medical therapy. These measures should include sun-protective measures, such as sunlight avoidance, the use of sun-protective clothing and sunscreen with sun protective factor (SPF) of 30 or higher. Pruritis due to skin disease can be disabling and can be managed by employing local agents (pramoxine, menthol, camphor) or oral drugs (e.g., sedating antihistamines, amitriptyline, gabapentin). Medical therapy for skin disease includes topical agents and systemic medications. Topical agents include corticosteroids and calcineurin inhibitors. Most patients end up requiring systemic drugs to control skin disease. The most commonly used systemic agents to treat skin disease are hydroxychloroquine and methotrexate. Although systemic glucocorticoids can control muscle disease, they are not effective in controlling skin disease.
Calcinosis, which occurs more frequently in juvenile dermatomyositis, can be managed with calcium channel blockers such as diltiazem. In some cases, surgical removal of calcinotic nodules may be indicated.
Physical therapy and rehabilitation play an essential role in management. Patients with mild disease should be encouraged to participate in active exercise programs. Range of motion exercises can help in preventing contractures. Patients with esophageal dysfunction may require consultation with speech therapy and may also require measures to prevent aspiration. Anti-aspiration measures include elevation of the head off the bed, thickening of feeds, and even feeding via gastric tubes when indicated.
Anti-resorptive therapy may be indicated in patients on long term systemic corticosteroids in order to prevent osteoporosis. Patients on high dose systemic glucocorticoids or immunosuppressants should be considered for prophylaxis against Pneumocystis Jirovecii with trimethoprim and sulfamethoxazole. Lastly, all patients should receive the appropriate immunizations prior to receiving immunosuppressants.
Differential Diagnosis
The following conditions can also present with muscle weakness and should be excluded by history, physical exam, and investigations before a definitive diagnosis of dermatomyositis can be made:
- Inclusion body myositis: unlike the symmetric and proximal involvement in dermatomyositis, muscle weakness in inclusion body myositis is usually asymmetric and involves distal muscles like the wrist and finger flexors. Muscle atrophy may be prominent during the examination, which is rare in dermatomyositis. A muscle biopsy will show the characteristic inclusion bodies. Unlike dermatomyositis, inclusion body myositis is refractory to treatment with corticosteroids.
- Drug-induced myopathy: Eliciting a drug history is essential to rule out drug-induced myopathies. The most common offending drugs are statins, alcohol, penicillamine, colchicine, glucocorticoids, zidovudine, and antimalarials. Drug-induced myopathy may cause mild myalgia, or it may even be severe enough to cause rhabdomyolysis. The absence of skin findings and muscle biopsy can differentiate this condition from dermatomyositis.
- Hypothyroidism: like dermatomyositis, hypothyroidism can present with proximal weakness and elevated muscle enzymes. The presence of other signs and symptoms of hypothyroidism, muscle biopsy, and serum thyroid-stimulating hormone levels can be used to differentiate this condition from dermatomyositis.
- Myasthenia gravis: Unlike dermatomyositis, myasthenia gravis predominantly causes muscle weakness of the ocular and bulbar muscles, is associated with anti-acetylcholine receptor antibodies, and does not cause elevation of muscle enzymes.
- Polymyalgia rheumatica: can present with pain and stiffness of the muscles around the shoulder and pelvic girdle. This condition can be differentiated from dermatomyositis by the presence of inflammatory markers, absence of elevated muscle enzymes, and normal muscle strength. Moreover, this condition mainly affects people more than 50 years of age.
Other differential diagnoses include muscular dystrophies, motor neuron disease, neuropathy, inherited metabolic myopathies, and myasthenia gravis.
The presence of characteristic skin findings in addition to symmetric muscle weakness should help to distinguish dermatomyositis from these conditions. Electromyography helps differentiate dermatomyositis from neuropathic causes of weakness. Muscle biopsy showing the hallmark pathological features of dermatomyositis also helps to exclude other causes.
Prognosis
The mortality rate of dermatomyositis is estimated to be 10% and is especially high in the first year of the disease.[28] The most common causes of death are malignancy, pulmonary complications, and ischemic heart disease. The following prognostic factors indicate higher mortality and poor outcome:
- Advanced age
- Initiation of treatment more than six months following the onset of symptoms
- Severe muscle weakness on presentation
- Presence of dysphagia
- Pulmonary involvement in the form of respiratory muscle weakness or interstitial lung disease
- Cardiac involvement
- Underlying malignancy[29][30][31][32]
Sixty-five percent of patients who survive have normal strength, 34% have a mild disability, and 16% have no disability. With treatment, 20% of patients attain remission, whereas 80% have a chronic or polycyclic course.[33]
Complications
Respiratory Disease
Pulmonary involvement may occur in the form of hypoventilation, aspiration pneumonia, or interstitial lung disease. Interstitial lung disease (ILD) is present in approximately a third of patients with dermatomyositis and is strongly associated with the presence of anti-histidyl transfer ribonucleic acid synthetase antibodies. Aspiration pneumonia can occur due to respiratory muscle weakness and can lead to significant morbidity and mortality.
Malignancy
Patients with dermatomyositis are at an increased risk of malignancies, which occur in 24% of cases.[34] A population-based study of patients from Sweden, Denmark, and Finland revealed a standardized incidence ratio of 3.0. Risk factors for malignancy included old age, absence of interstitial lung disease, severe cutaneous involvement (or shawl sign), presence of anti-155/140 or anti-NXP2 antibodies, absence of myositis specific antibodies, resistance to treatment, and history of previous malignancy with relapse. The most common malignancies were adenocarcinomas of ovary, lung, pancreas, stomach, and colon, as well as non-Hodgkin lymphoma [35]. The risk of malignancy was highest in the first year of the disease and remains high for up to five years. Even after this period, the risk of malignancy was higher than that found in the general population[36].
Heart Disease
Cardiac involvement in dermatomyositis is usually subclinical. Electrocardiography (ECG) may reveal conduction abnormalities and arrhythmias. Cardiac involvement may occur in the form of myocarditis, congestive heart failure, or coronary artery disease.[37]
Esophageal Disease
Patients can develop dysphagia due to oropharyngeal and esophageal muscle weakness. These conditions can lead to malnutrition and can also increase the risk of aspiration.
Other complications may also include calcinosis, muscle atrophy, and contractures.
Deterrence and Patient Education
Patients and their families should be educated about the nature of the disease, its progression, prognosis, and the need to monitor for adverse effects of medical therapy. In addition, the following measures are recommended:
Sun Protective Measures
Patients should be advised to avoid sunlight, use sunscreen, and adopt sun-protective measures such as wearing wide-brimmed hats and full-body covered clothing to prevent the worsening of photosensitive skin rash.
Diet
Patients with severe muscle inflammation may require a high protein diet. The presence of esophageal dysfunction may require a special diet and an elevation of the head of the bed after meals.
Physical Activity
Physical exercise and rehabilitation is an important part of dermatomyositis management. Aerobic exercise and resistance training help to enhance muscle strength. Range of motion exercise is also helpful to prevent contractures. If muscle inflammation is severe, sustained rest should be advised, and physical activity should be avoided.
Enhancing Healthcare Team Outcomes
Dermatomyositis is a rare condition and is best managed by a multidisciplinary team and may even require referral to a tertiary care center. All physicians must be able to recognize the typical presentation of dermatomyositis as most patients initially present to a rheumatologist, internist, family physician, or pediatrician, depending on their age.
The recently formulated European League Against Rheumatism and the American College of Rheumatology (EULAR/ACR) criteria for idiopathic inflammatory myopathies is a highly sensitive and specific probability-based classification system which can be used to identify cases of dermatomyositis. Initial workup for patients with suspected dermatomyositis includes testing for muscle enzymes and myositis specific autoantibodies. Electromyography, muscle, and skin biopsy may be done if the diagnosis is uncertain, especially in patients who lack characteristic findings. In addition to a comprehensive evaluation and assessment for malignancy, consultations with other specialties may be indicated depending on the type of associated complications. For example, patients with underlying malignancy should be referred to oncology. The presence of extra muscular manifestations warrants consultations with pulmonology, gastroenterology, cardiology, and speech therapy. Patients with a skin rash should be referred to a dermatologist for assessment, management, and follow up. Early recognition and prompt initiation of treatment can reduce the morbidity and mortality of patients. The first-line treatment for dermatomyositis is prednisolone.[38] [Level 5]
Concomitant treatment with glucocorticoids and immunosuppressants should be given to patients who are resistant to treatment with glucocorticoids alone or have extra muscular complications or severe weakness. Sun avoidance and sun-protective measures are recommended in all patients with skin disease. Rehabilitation in the form of resistive exercises should be started in the early phase of the disease as it can improve daily functioning.[39] [Level 5] Patients should be followed up at regular intervals to follow the course of the disease and to monitor for response to therapy, adverse effects of therapy, systemic complications, and malignancy.