Continuing Education Activity
Bilirubin circulates in both the unconjugated and conjugated forms. Disorders of impaired conjugation of bilirubin classically present with elevated levels of unconjugated bilirubin, also called indirect bilirubin. Major causes of impaired conjugation of bilirubin include inherited disorders and secondary causes such as drugs. Disorders can range from asymptomatic to severe bilirubin encephalopathy, which is also known as kernicterus. This activity reviews inherited disorders of impaired bilirubin conjugation and the role of the interprofessional team in their management.
Objectives:
Determine the etiologies of impaired bilirubin conjugation.
Identify the presenting features of impaired bilirubin conjugation.
Assess the management options for impaired bilirubin conjugation.
Communicate the need for an interprofessional team approach to caring for a patient with impaired bilirubin conjugation.
Introduction
Bilirubin circulates in both the conjugated and unconjugated forms (predominant). Disorders of impaired bilirubin conjugation classically present with elevated levels of unconjugated or indirect bilirubin.[1][2] Major causes of impaired conjugation of bilirubin include inherited disorders and secondary causes such as drugs.[3][4] Disorders can range from an asymptomatic presentation to a very severe bilirubin encephalopathy (kernicterus). In this review, we focus on the inherited disorders and their management.
Etiology
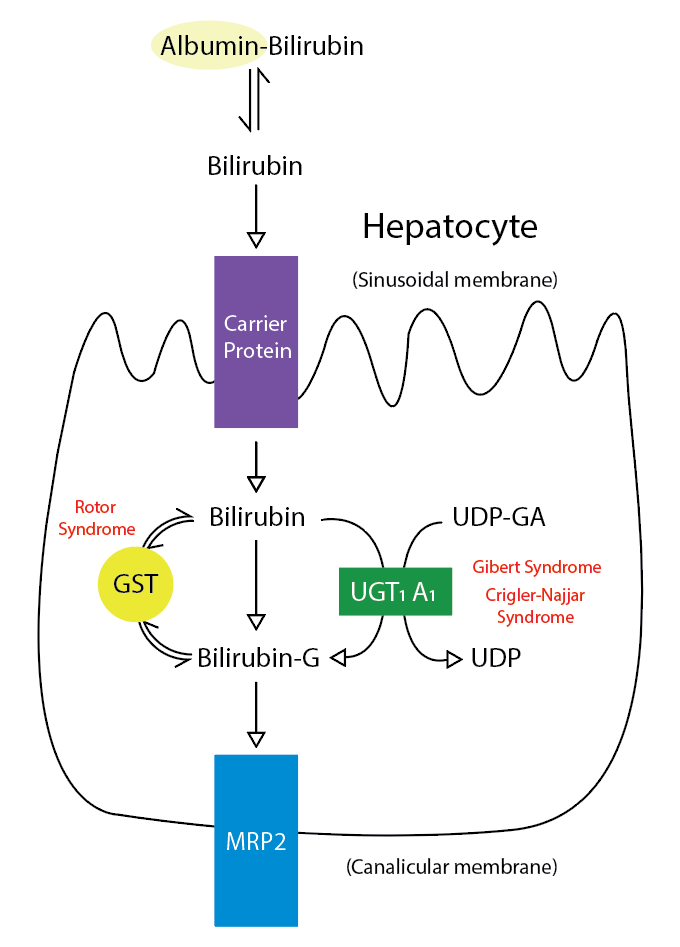
Seventy percent to 90% of bilirubin is derived from red blood cell heme.[5][6][7] Heme oxygenase converts this heme to biliverdin, which is converted by biliverdin reductase to unconjugated bilirubin (UCB). UCB bound to albumin enters the hepatocyte, where it binds glutathione-S-transferases superfamily members, which present it for conjugation. The process of conjugation occurs in two steps. Bilirubin monoglucuronide initially forms through the transfer of a glucuronic acid molecule to a carboxyl group of bilirubin through the enzyme UDP-glucuronosyl transferase (UGTA1A), which is located in the endoplasmic reticulum. One more glucuronic acid molecule is added to a second carboxyl group of bilirubin to form bilirubin diglucuronide. This overall process allows bilirubin to become water-soluble for easier transport. Bilirubin diglucuronide can be excreted into bile or secreted into the plasma for reuptake. Disorders of impaired bilirubin conjugation fundamentally arise from the impaired activity of the UGTA1A enzyme. Gilbert syndrome and Crigler-Najjar syndrome have inherited diseases that cause impaired bilirubin conjugation. Lucy-Driscoll syndrome is also a rare inherited disorder that can potentially contribute to severe transient familial unconjugated hyperbilirubinemia in an infant’s early life. Infants and mothers with this syndrome contain an unknown factor that inhibits UGTA1A enzyme activity. Unconjugated hyperbilirubinemia can also manifest as neonatal jaundice because of immature UGTA1A enzyme activity and increased breakdown of fetal erythrocytes. Protease inhibitors, pregnanediol, novobiocin, chloramphenicol, and gentamycin, are common drugs that inhibit the UGTA1A enzyme (See Image. Metabolic Pathway for Bilirubin in the Hepatocyte).
Epidemiology
Crigler-Najjar syndrome is extremely rare, with an occurrence of around 1 case per million live births. The prevalence of Gilbert syndrome in the United States is much higher at 5% to 10%, and afflicts people predominantly of European descent. In Gilbert syndrome, there are differences in the mutation site of the UGTA1A according to ethnic group. For instance, white populations with Gilbert syndrome most commonly have a mutation in the TATAA element of the promoter region.
Pathophysiology
Gilbert syndrome is an autosomal recessive disorder that presents most commonly due to adding an extra dinucleotide sequence, TA, to the TATA promoter in the UGTA1A gene. UGTA1 enzyme activity is 10% to 30% of its normal activity and presents with mild unconjugated hyperbilirubinemia with bilirubin levels less than 5 mg/dl. Jaundice occurs during stress or fasting when bilirubin levels exceed 3 mg/dl. Crigler-Najjar type 1 syndrome is an autosomal recessive disorder that occurs most commonly due to mutations in exons 2 to 5 of UGT1A1, leading to complete or nearly complete loss of enzyme activity. Elevated unconjugated bilirubin levels between 20 to 45 mg/dl appear after birth, and life-threatening symptoms such as bilirubin encephalopathy (Kernicterus) arise. Crigler-Najjar type 2 syndrome, also an autosomal recessive disorder, is the much milder variant of this disease due to UGT1A1 functioning at less than 10% of its capacity and presents with rare kernicterus during times of stress, fasting, or illness. A bilirubin level greater than 15 mg/dl appears to increase the kernicterus risk. Drugs such as protease inhibitors like Indinavir can also cause disorders of bilirubin metabolism by competitively inhibiting the UGT1A1 enzyme.
History and Physical
Gilbert syndrome presents in patients as mild, intermittent, and prolonged unconjugated hyperbilirubinemia without hemolysis. It also presents normal LDH, haptoglobin levels, reticulocyte counts, and normal liver function tests. The liver ultrasound is normal. Gilbert syndrome is most commonly asymptomatic, and it is often found incidentally on routine blood testing conducted for other reasons. Upon physical exam, this syndrome can manifest as jaundice. Nonspecific symptoms of abdominal cramps, fatigue, and malaise may also present with the disease. A thorough history should be taken to ascertain whether the patient is taking drugs that are metabolized through glucuronidation, such as tolbutamide, rifamycin, HIV protease inhibitors, gemfibrozil, and statins, which could contribute to drug toxicity.
Crigler-Najjar type 1 syndrome presents with jaundice in infants several days after they are born. This jaundice becomes rapidly more severe in the second week of the infant’s life. Patients have the potential to develop kernicterus and show symptoms such as extrapyramidal dystonia, hearing loss, and oculomotor paresis. Crigler-Najjar type 2 syndrome is most often asymptomatic but can present with jaundice and kernicterus during times of stress, illness, or fasting when bilirubin levels exceed more than 15 mg/dl.
Evaluation
Diagnosis of Gilbert syndrome can be suspected through the detection of hyperbilirubinemia on routine blood tests with an increase in unconjugated bilirubin. It is important to follow up with the patient with tests to exclude liver disease and hemolysis for at least 6 months before making the diagnosis. Detecting the A(TA)7TAA genotype through genetic testing can definitively diagnose the syndrome. However, this technique should only be used when the serum bilirubin level is greater than 5.0 mg/dl or treatment with irinotecan will be utilized. The diagnosis can be confirmed by repeating bilirubin levels before and after prolonged fasting, exacerbating the increase in unconjugated bilirubin.[8] However, this is rarely required.
There are no simple tests to diagnose Crigler Najjar syndrome type 1. Genetic testing can be utilized to sequence the coding region for known mutations, such as exons 2 to 5 in UGT1A1, and make a definitive diagnosis. However, multiple tests can be used to differentiate between Crigler Najjar syndrome type 1 and type 2. Lowered levels of serum bilirubin, usually less than 15 mg/dL, and a greater than 30% decrease in serum bilirubin after administration of phenobarbital indicate a diagnosis of Crigler Najjar syndrome type 2.[9] There is no effect of phenobarbital on the severe enzymopathy of type 1 syndrome. Bile pigment analysis of bilirubin conjugates will also show very low levels of conjugated bilirubin in type 1 and high levels of mono-conjugated bilirubin in type 2.
Treatment / Management
When a patient is diagnosed with Gilbert syndrome, they should be informed of the condition’s benign nature. Treatment is not required for Gilbert syndrome. However, if the patient’s jaundice becomes severe enough, microsomal enzyme inducers such as phenobarbital can be prescribed. The patient should also be educated about the cautious use of drugs metabolized by glucuronidation, such as statins, protease inhibitors, and especially Irinotecan. Irinotecan can cause severe symptoms such as diarrhea and myelosuppression because of impaired clearance in patients with Gilbert syndrome. Accordingly, patient education and careful monitoring can potentially prevent these sorts of severe drug side effects.
Treatment and management of Crigler Najjar syndrome type 1 revolve around controlling serum bilirubin levels below the threshold for kernicterus, 10 mg/dl. This is accomplished through exchange transfusions and phototherapy for 8 to 12 hours daily. Phototherapy, though its effectiveness decreases over time, converts bilirubin into water-soluble forms that can be secreted into bile and excreted in the urine without conjugation. Moreover, liver transplantation is the most effective and definitive form of treatment. Earlier intervention through transplantation decreases the risk and magnitude of brain damage. Successful transplantation can reverse brain damage if it is not too severe. Gene therapy could be the most prominent therapy, given the recent success with other severe genetic disorders. Because Crigler Najjar syndrome type 2 is much less severe, lifelong phenobarbital therapy is sufficient. By inducing UGT1A1, Phenobarbital can reduce the bilirubin concentration by more than 25%.
Differential Diagnosis
Given a presentation of isolated unconjugated hyperbilirubinemia, hemolytic disorders such as sickle cell anemia, pyruvate kinase, and G6PD deficiencies need to be excluded. Disorders such as hepatic failure and portosystemic shunts can also contribute to impaired hepatic bilirubin uptake and should be considered, though the clinical presentation of these particular disorders will be obvious. It should be noted that in addition to inherited disorders and particular medication side effects, advanced cirrhosis, hyperthyroidism, and immature conjugating ability, which is often seen in neonates, could cause impaired bilirubin conjugation.
Prognosis
The prognosis of Gilbert syndrome is excellent. A patient with this disorder can live a normal life for the most part. Phenobarbital usage is only indicated when hyperbilirubinemia causes severe jaundice. Crigler Najjar syndrome type 1 presents with a poor prognosis because of the high risk of kernicterus. The prognosis of the type 2 variant is much more favorable.
Pearls and Other Issues
Despite its inconvenient nature, Gilbert syndrome may have a protective effect regarding cardiovascular disease, given that bilirubin is a potent antioxidant. Studies have illustrated an inverse relationship between serum bilirubin levels and coronary artery calcification and ischemic stroke.
Enhancing Healthcare Team Outcomes
The management of unconjugated hyperbilirubinemia requires an interprofessional approach as the differential diagnosis is large. The primary care physician, nurse practitioner, internist, pediatrician, and gastroenterologist will most likely encounter patients with Gilbert and Crigler Najjar syndrome. The former is a benign condition that requires no treatment. The key is patient education and avoiding medications that interfere with liver function. Crigler Najjar syndrome is managed in multiple ways, including exchange transfusion, phototherapy, and sometimes liver transplantation. Gene therapy could be the most prominent therapy, given the recent success with other severe genetic disorders. Because Crigler Najjar syndrome type 2 is much less severe, lifelong phenobarbital therapy is sufficient. By inducing UGT1A1, Phenobarbital can reduce the bilirubin concentration by more than 25%. The outcomes for Gilbert syndrome are excellent, but for patients with Crigler Najjar syndrome, the outcomes are fair to guarded.[4][9]