Continuing Education Activity
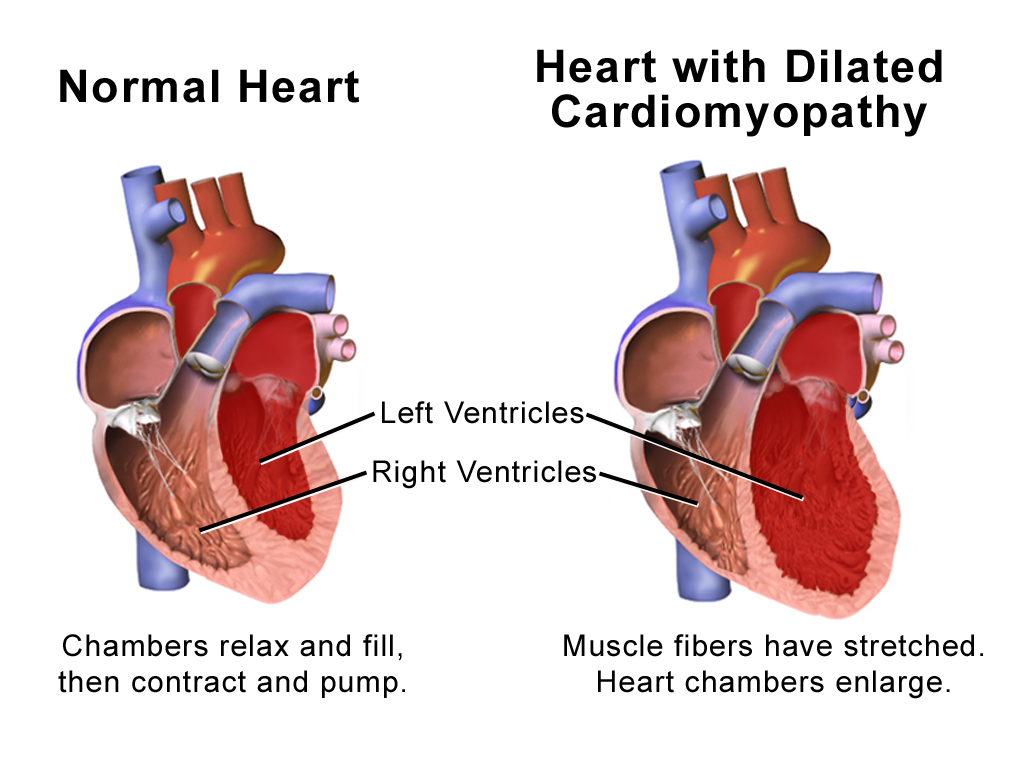
Dilated Cardiomyopathy (DCM) is a disease of the heart muscle characterized by enlargement and dilation of one or both of the ventricles along with impaired contractility defined as left ventricular ejection fraction (LVEF) less than 40%. By definition, patients have systolic dysfunction and may or may not have overt symptoms of heart failure. This disease process can be classified as either primary or secondary DCM. Primary DCM is considered idiopathic and the diagnosis can only be made after excluding secondary causes. This activity reviews the causes of dilated cardiomyopathy, its presentation, and highlights the role of the interprofessional team in its management.
Objectives:
- Describe the causes of dilated cardiomyopathy.
- Recall the presentation of dilated cardiomyopathy.
- Summarize the treatment options for dilated cardiomyopathy.
- Review the importance of improving care coordination among interprofessional team members to improve outcomes for patients affected by dilated cardiomyopathy
Introduction
Dilated Cardiomyopathy (DCM) is a disease of the heart muscle characterized by enlargement and dilation of one or both of the ventricles along with impaired contractility defined as left ventricular ejection fraction (LVEF) less than 40%. By definition, patients have systolic dysfunction and may or may not have overt symptoms of heart failure. This disease process can be classified as either primary or secondary DCM. Primary DCM is considered idiopathic and the diagnosis can only be made after excluding secondary causes.[1][2][3]
In most cases DCM is progressive, leading to heart failure and death. Without a transplant, the survival rates are poor.
DCM has many causes and all of them affect the ventricular function to a varying degree. While most patients with DCM have symptoms, a few patients may be asymptomatic because of the compensatory mechanisms. The continued enlargement fo the ventricles leads to a decline in ventricular function, followed by conduction system abnormalities, ventricular arrhythmias, thromboembolism, and heart failure. The earlier these patients are identified and treated, the better the prognosis.
Etiology
The most common etiology of dilated cardiomyopathy (DCM) is idiopathic and without an identifiable cause. DCM can have a familial or genetic predisposition although these cases are usually classified under idiopathic if no clear genetic link is identified. DCM has been associated with mutations in genes for Desmin (cytoskeletal), Lamin C (nuclear membrane), or Myosin (contractile proteins). The secondary causes include infectious myocarditis (e.g., viral, Chagas disease, Lyme disease), ischemic disease, hypertension, medication-induced (e.g., Anthracyclines), alcohol abuse, human immunodeficiency virus (HIV), peripartum cardiomyopathy, or infiltrative disease. Ischemic cardiomyopathy caused by coronary artery disease (CAD) is the most common cause of congestive heart failure. However, ischemic cardiomyopathy is classified as its own disease entity and is only described as a cause of DCM in occult disease in patients without known CAD. Stress cardiomyopathy, also known as Takotsubo cardiomyopathy or Broken heart syndrome, is a relatively uncommon but increasingly reported cause. However, it is often classified as its own entity separate from primary DCM. It is characterized by transient ballooning of the left ventricular (LV) apex typically following a severe psychological or physiological stress that is believed to be secondary to intense catecholamine surge.[4]
Epidemiology
DCM is more commonly seen in men than in women. Its prevalence in the general population is estimated at 36 cases per 100,000. DCM accounts for 10,000 deaths and 46,000 hospitalizations in the United States annually. These figures may underestimate the true prevalence because many patients are asymptomatic and, therefore, undiagnosed despite LV dysfunction. [5][2]
Pathophysiology
Many cases of dilated cardiomyopathy (DCM) are due to idiopathic etiology. But, it also can arise from various myocardial insults. Enlargement of the ventricles can either be secondary to LV failure or secondary to a primary cardiomyopathic process and can be associated with both systolic and diastolic dysfunction. Reduction in systolic function is believed to be caused by myocardial remodeling that results in an increase in both end-systolic and end-diastolic volumes.
The progressive dilatation of the ventricles leads to significant tricuspid and mitral valve insufficiency, which further lower the ejection fraction and increase the ventricular wall stress and end systolic volumes. Early compensatory mechanisms include an increase in heart rate and tone of the peripheral vascular system. However, these compensatory mechanisms lead to geometric remodeling of the ventricles and eventually this leads to worsening of the myocardial injury. At the same time, there is neurohumoral activation of the renin-angiotensin aldosterone system and an increase in circulating levels of catecholamines. In addition, levels of natriuretic peptides are also increased. Eventually these compensatory mechanisms become overwhelmed and the heart fails.
Histologic examination or the myocardium typically shows nonspecific changes of fibrosis and hypertrophy. It also reveals myocardial injury with a marked infiltrate by inflammatory cells. [6]
History and Physical
The majority of cases of dilated cardiomyopathy (DCM) present between the ages of 20 and 60; however, DCM can be seen in children or the elderly. A large number of patients with DCM may have a long latent period where they are clinically asymptomatic. When symptoms do arise, they are the result of LV systolic dysfunction. In addition to a focused cardiac history and examination, a more thorough evaluation is recommended to identify any systemic disease or secondary causes.
Classic symptoms include paroxysmal nocturnal dyspnea, orthopnea, leg swelling, and shortness of breath. Nonspecific symptoms of fatigue, malaise, and weakness also can be present. More severe cases can present with thromboembolic complications, conduction disturbances, arrhythmias or even sudden cardiac death. Physical examination findings are largely not specific to other causes of cardiomyopathy and consist of typical findings seen with congestive heart failure.
Findings include crackles in the lung fields, elevated jugular venous pressures, peripheral edema, and an S3 gallop. Classically, the point of maximum impulse or PMI is displaced laterally. Tricuspid or mitral regurgitation murmurs are not uncommon as a result of ventricular enlargement and annular dilation. Neck examination may reveal jugular venous distension, A-wave, large V waves, and positive hepatojugular reflux.
Evaluation
Evaluation for secondary causes of dilated cardiomyopathy (DCM) always should be pursued prior to making the diagnosis of idiopathic DCM. Workup is focused on identifying any possible reversible causes. Recommended laboratory testing includes thyroid function tests, HIV serology, electrolytes, and iron studies (to rule out hemochromatosis). Urine toxicology screen and alcohol level can be checked when substance abuse is suspected. In certain familial cases, genetic testing should be considered. Serum B-type natriuretic peptide (BNP) levels may be obtained in cases where the diagnosis is unclear. Low levels of BNP are useful in ruling out CHF. In addition, levels of BNP are useful for prognosis.
One should also rule out hypothyroidism and anemia.
Chest X-ray may show cardiomegaly and evidence of pulmonary effusions and venous congestion. Electrocardiogram (EKG) may show nonspecific ST segment and T wave abnormalities. ECG may also reveal atrial fibrillation.
Oxygen consumption per minute of less than 14 ml/kg/min indicates a poor prognosis.
Echocardiography is crucial in making the diagnosis of DCM and provides an objective assessment of ventricular size, function, and any associated valvular abnormalities. Echocardiography also can identify the presence of a mural thrombus. Finally, echocardiography can help in differentiating DCM from hypertrophic and restrictive cardiomyopathy.
Coronary angiography should be performed in those without a known history of CAD to further define coronary anatomy and rule out occult ischemic disease as the cause of DCM. Very rarely, a myocardial biopsy is needed for the evaluation of storage diseases or infiltrative causes when suspected. As mentioned, histologic findings of idiopathic DCM are nonspecific, and biopsy exposes patients to unnecessary risk.[7]
Treatment / Management
Besides treating any identifiable and reversible underlying causes, the management and treatment of Dilated Cardiomyopathy (DCM) are in concordance with the standard heart failure guidelines.
In patients with acute congestive heart failure exacerbation, intravenous loop diuretics are given to treat hypervolemia. Management of chronic and stable disease with oral diuretics often is needed to achieve a euvolemic state. Angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARB) have shown benefit in the treatment of heart failure with reduced ejection fraction and are recommended in patients with DCM. Aldosterone receptor blockade with spironolactone or eplerenone also is recommended in patients with New York Heart Association (NYHA) heart failure class II-IV and systolic dysfunction. Similarly, beta-blockade with carvedilol, bisoprolol, or long-acting metoprolol is recommended in all patients with heart failure with reduced ejection fraction without any contraindications. The addition of isosorbide dinitrate plus hydralazine also has shown to increase survival amongst those with advanced disease.
Anticoagulation should be used for patients with artificial valves, atrial fibrillation, and known mural thrombus. Oral anticoagulants can reduce the risk of stroke, but the risk of bleeding is always present.
Finally, patients with disease refractory to maximum medical therapy should be considered for cardiac transplantation and LVAD as a bridge or for "destination" therapy in those who are not candidates for transplantation. Implanted cardioverter defibrillators (ICD) for primary prevention of sudden cardiac death and cardiac resynchronization therapy (CRT) can be considered and are recommended by the heart failure guidelines.[8][9][10]
A heart transplant is an option but the lack of donors is a major stumbling block. Indications for a heart transplant include refractory cardiogenic shock, ventricular arrhythmias, dependence on high levels of inotropes and dependency on an IABP or a ventricular assist device.
Differential Diagnosis
- Cardiac tamponade
- Acute pericarditis
- Hypertrophic cardiomyopathy
- Restrictive cardiomyopathy
Staging
NYHA Classification of heart failure
- Stage A: Having risk factors for developing CHF including hypertension, diabetes, CAD, and a family history of cardiomyopathy
- Stage B: Asymptomatic heart failure; having left ventricular systolic dysfunction, prior MI, asymptomatic valvular disease
- Stage C: Symptomatic heart failure: having dyspnea, reduced exercise tolerance, and fatigue
- Stage D: Refractory end-stage heart failure with symptoms at rest despite maximal medical therapy and recurrent admissions
Prognosis
Overall, the prognosis of patients with dilated cardiomyopathy is guarded. Most patients eventually end up with chronic heart failure. Many become candidates for a heart transplant or an assist device which also adds more morbidity. The progression to heart failure depends on the ejection fraction and cause of the disease. Almost 50% of patients are dead within 5 years. Negative prognostic factors include advanced NYHA classification, male sex, severe CHF, and renal failure. Individuals who have symptoms at rest and/or unable to exercise usually have the poorest prognosis. Peak V02 levels are now often used to predict mortality; those with high V02 levels tend to have a far better prognosis than those with low levels. With optimal medical therapy, patients with mild CHF can have a reasonably good quality of life.
Complications
- Congestive heart failure
- Cerebrovascular accident
- Valvular heart disease
- Abnormal cardiac rhythms
- Sudden cardiac death
- Thromboembolism
Postoperative and Rehabilitation Care
All patients with dilated cardiomyopathy need intense education on the diet. Both salt and water restrictions are necessary to prevent symptoms. Patients should remain physically active or enroll in a cardiac rehab program. Regular cardiac rehabilitation can lower the mortality rate by 20%, relieve symptoms and reduce adverse cardiac events.
Consultations
- Cardiac surgeon
- Cardiologist
- Critical care specialist
- Dietitian
Deterrence and Patient Education
Patient education regarding medication compliance, dietary restrictions and regular follow-up, is critical for management of dilated cardiomyopathy.
Pearls and Other Issues
- Unlike the past when all patients with DCM were empirically treated with anticoagulation, today the guidelines recommend the use of anticoagulants only in patients with atrial fibrillation, prosthetic heart valves, or known mural thrombus.
- When there is progressive end-stage heart failure despite maximal medical therapy and the prognosis is deemed poor, one may consider a heart transplant.
- All patients with DCM must be educated on the disorder and the importance of dietary restrictions in sodium and water.
- Patients with DCM should be referred for cardiac rehabilitation as this has been shown to reduce all-cause mortality by 20-30% over 5 years, including improvement in symptoms.
Enhancing Healthcare Team Outcomes
Dilated cardiomyopathy is a progressive heart disorder with no cure. Eventually, most patients progress to heart failure and close to 50% are dead within five years. Today many treatments have been devised for the treatment of dilated cardiomyopathy, but for most of them, there is a lack of evidence to support their use. There is no longer any question that this disorder is best managed by an interprofessional team that specializes in heart disease. The three single most important factors that determine the prognosis are 1) diet, 2) medication compliance, and 3) an exercise program. The disorder should be risk-stratified and managed accordingly.[11][12]
A dietitian or a nurse is vital because one of the best ways to prevent progression to heart failure is by restricting fluid and salt intake. The family must be told to weigh the patient and offer foods that are low in salt. The pharmacist must ensure that the patient remains compliant with the medications. There is ample evidence indicating that participating in an exercise program can reduce symptoms and lower all-cause mortality. If the patient is deemed to be a candidate for a heart transplant, the heart transplant team should be consulted early on. Close communication between the team is vital if one wants to improve outcomes.
Outcomes
The outcomes of patients with dilated cardiomyopathy depend on the cause, ejection fraction, and comorbidity. At least 50% are dead within five years, and many others go on to develop heart failure. Despite many treatments available, the majority remain experimental. In view of the lack of data on treatments, it is imperative that the education of the patient and family be done to ensure that they comply with fluid and salt restriction. [13][14]