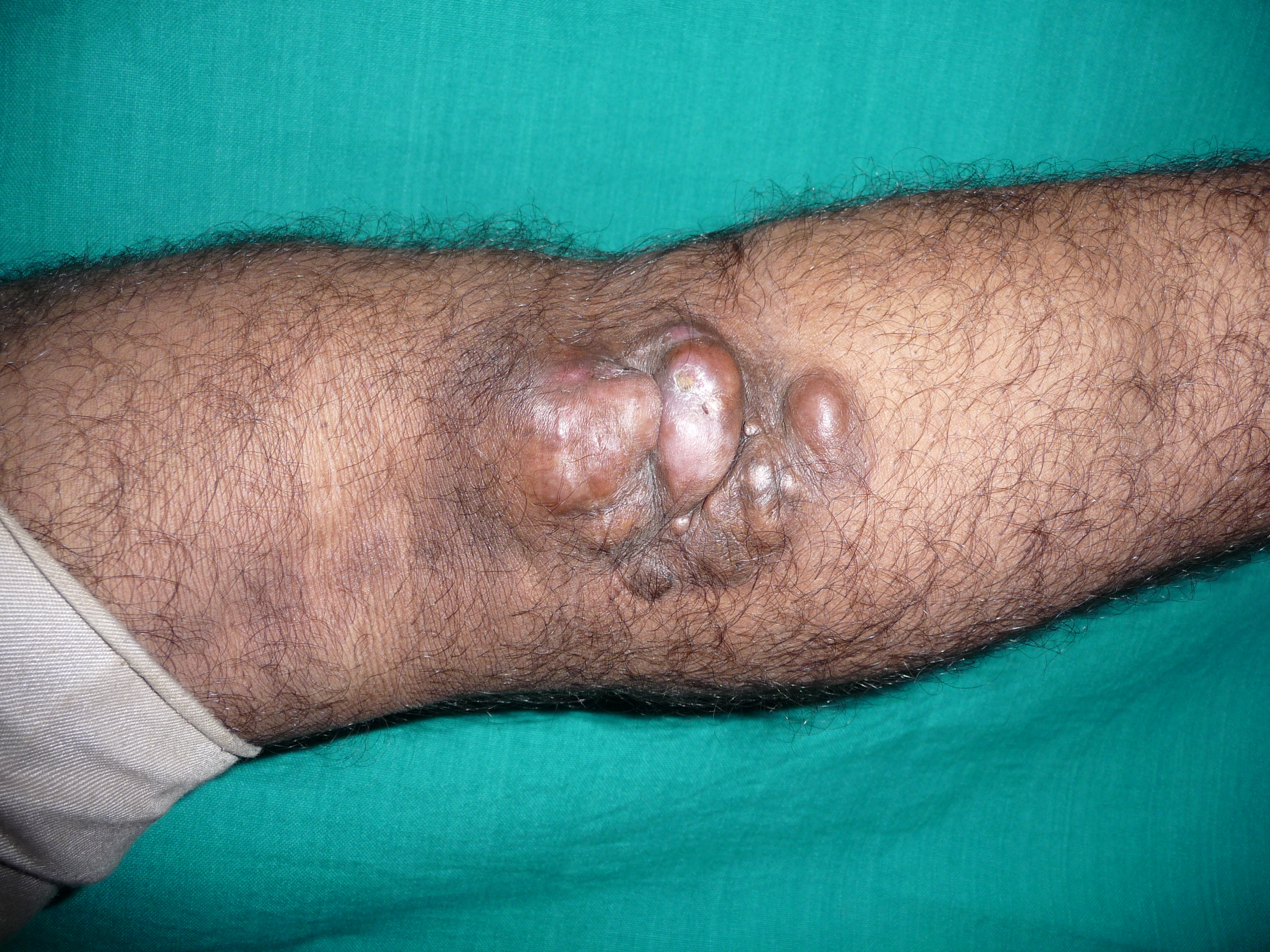
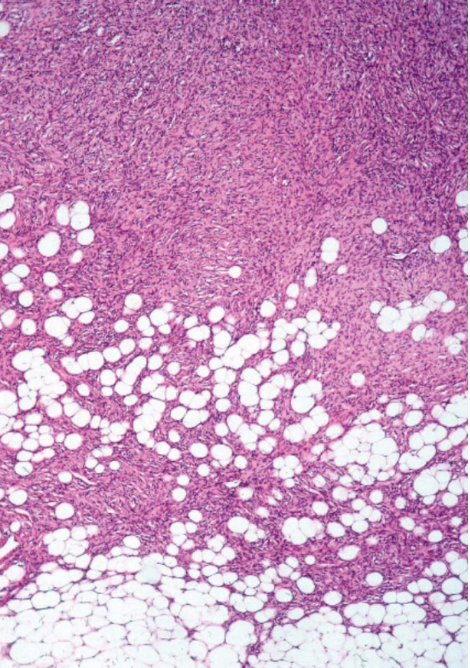
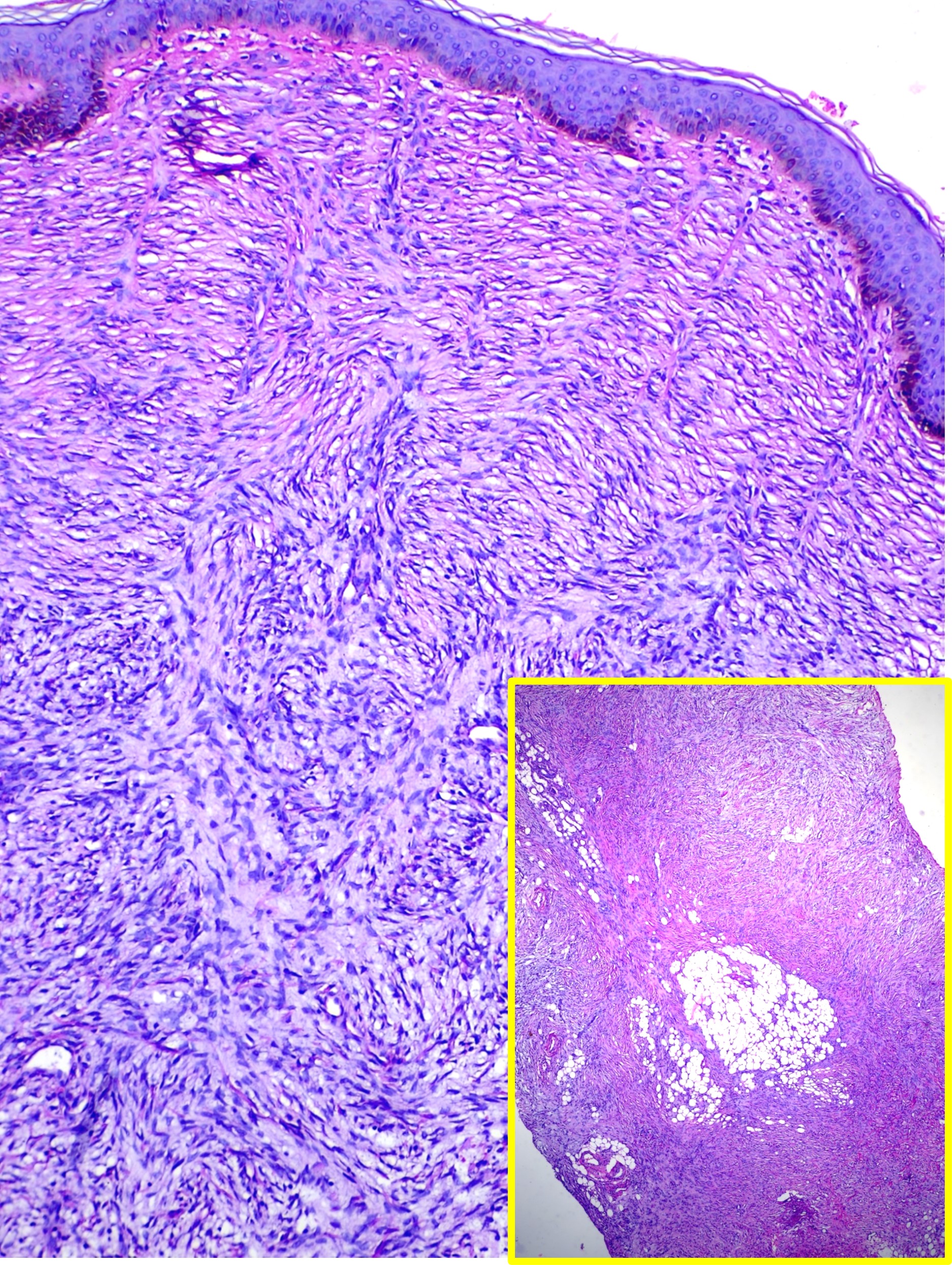
[1]
Rutkowski P, Van Glabbeke M, Rankin CJ, Ruka W, Rubin BP, Debiec-Rychter M, Lazar A, Gelderblom H, Sciot R, Lopez-Terrada D, Hohenberger P, van Oosterom AT, Schuetze SM, European Organisation for Research and Treatment of Cancer Soft Tissue/Bone Sarcoma Group, Southwest Oncology Group. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010 Apr 1:28(10):1772-9. doi: 10.1200/JCO.2009.25.7899. Epub 2010 Mar 1
[PubMed PMID: 20194851]
Level 1 (high-level) evidence
[2]
Criscito MC, Martires KJ, Stein JA. Prognostic Factors, Treatment, and Survival in Dermatofibrosarcoma Protuberans. JAMA dermatology. 2016 Dec 1:152(12):1365-1371. doi: 10.1001/jamadermatol.2016.1886. Epub
[PubMed PMID: 27262160]
[3]
Trofymenko O, Bordeaux JS, Zeitouni NC. Survival in patients with primary dermatofibrosarcoma protuberans: National Cancer Database analysis. Journal of the American Academy of Dermatology. 2018 Jun:78(6):1125-1134. doi: 10.1016/j.jaad.2017.11.030. Epub 2017 Nov 23
[PubMed PMID: 29175214]
[4]
Martin L, Piette F, Blanc P, Mortier L, Avril MF, Delaunay MM, Dréno B, Granel F, Mantoux F, Aubin F, Sassolas B, Adamski H, Dalac S, Pauwels C, Dompmartin A, Lok C, Estève E, Guillot B, French Group for Cutaneous Oncology. Clinical variants of the preprotuberant stage of dermatofibrosarcoma protuberans. The British journal of dermatology. 2005 Nov:153(5):932-6
[PubMed PMID: 16225602]
[5]
Takahira T, Oda Y, Tamiya S, Higaki K, Yamamoto H, Kobayashi C, Izumi T, Tateishi N, Iwamoto Y, Tsuneyoshi M. Detection of COL1A1-PDGFB fusion transcripts and PDGFB/PDGFRB mRNA expression in dermatofibrosarcoma protuberans. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2007 Jun:20(6):668-75
[PubMed PMID: 17431412]
[6]
Simon MP, Pedeutour F, Sirvent N, Grosgeorge J, Minoletti F, Coindre JM, Terrier-Lacombe MJ, Mandahl N, Craver RD, Blin N, Sozzi G, Turc-Carel C, O'Brien KP, Kedra D, Fransson I, Guilbaud C, Dumanski JP. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nature genetics. 1997 Jan:15(1):95-8
[PubMed PMID: 8988177]
[7]
Haycox CL, Odland PB, Olbricht SM, Piepkorn M. Immunohistochemical characterization of dermatofibrosarcoma protuberans with practical applications for diagnosis and treatment. Journal of the American Academy of Dermatology. 1997 Sep:37(3 Pt 1):438-44
[PubMed PMID: 9308560]
[8]
Allen A, Ahn C, Sangüeza OP. Dermatofibrosarcoma Protuberans. Dermatologic clinics. 2019 Oct:37(4):483-488. doi: 10.1016/j.det.2019.05.006. Epub
[PubMed PMID: 31466588]
[9]
Larbcharoensub N, Kayankarnnavee J, Sanpaphant S, Kiranantawat K, Wirojtananugoon C, Sirikulchayanonta V. Clinicopathological features of dermatofibrosarcoma protuberans. Oncology letters. 2016 Jan:11(1):661-667
[PubMed PMID: 26870263]
Level 3 (low-level) evidence
[10]
Thway K, Noujaim J, Jones RL, Fisher C. Dermatofibrosarcoma protuberans: pathology, genetics, and potential therapeutic strategies. Annals of diagnostic pathology. 2016 Dec:25():64-71. doi: 10.1016/j.anndiagpath.2016.09.013. Epub 2016 Sep 27
[PubMed PMID: 27806849]
[11]
Li Y, Wang C, Xiang B, Chen S, Li L, Ji Y. Clinical Features, Pathological Findings and Treatment of Recurrent Dermatofibrosarcoma Protuberans. Journal of Cancer. 2017:8(7):1319-1323. doi: 10.7150/jca.17988. Epub 2017 May 12
[PubMed PMID: 28607608]
[12]
PDQ Pediatric Treatment Editorial Board. Childhood Soft Tissue Sarcoma Treatment (PDQ®): Health Professional Version. PDQ Cancer Information Summaries. 2002:():
[PubMed PMID: 26389361]
[13]
Llombart B, Serra C, Requena C, Alsina M, Morgado-Carrasco D, Través V, Sanmartín O. Guidelines for Diagnosis and Treatment of Cutaneous Sarcomas: Dermatofibrosarcoma Protuberans. Actas dermo-sifiliograficas. 2018 Dec:109(10):868-877. doi: 10.1016/j.ad.2018.05.006. Epub 2018 Jul 4
[PubMed PMID: 30539729]
[14]
Bhatt MD, Nambudiri VE. Cutaneous Sarcomas. Hematology/oncology clinics of North America. 2019 Feb:33(1):87-101. doi: 10.1016/j.hoc.2018.08.007. Epub
[PubMed PMID: 30497679]
[15]
Penel N, El Bedoui S, Robin YM, Decanter G. [Dermatofibrosarcoma: Management]. Bulletin du cancer. 2018 Nov:105(11):1094-1101. doi: 10.1016/j.bulcan.2018.08.008. Epub 2018 Oct 5
[PubMed PMID: 30297237]
[16]
Chappell AG, Doe SC, Worley B, Yoo SS, Gerami P, Alam M, Buck DW 2nd, Kim JYS, Wayne JD. Multidisciplinary surgical treatment approach for dermatofibrosarcoma protuberans: an update. Archives of dermatological research. 2021 Jul:313(5):367-372. doi: 10.1007/s00403-020-02124-8. Epub 2020 Aug 7
[PubMed PMID: 32770258]
[17]
Lyu A, Wang Q. Dermatofibrosarcoma protuberans: A clinical analysis. Oncology letters. 2018 Aug:16(2):1855-1862. doi: 10.3892/ol.2018.8802. Epub 2018 May 24
[PubMed PMID: 30008876]
Level 3 (low-level) evidence
[18]
Meguerditchian AN, Wang J, Lema B, Kraybill WG, Zeitouni NC, Kane JM 3rd. Wide excision or Mohs micrographic surgery for the treatment of primary dermatofibrosarcoma protuberans. American journal of clinical oncology. 2010 Jun:33(3):300-3. doi: 10.1097/COC.0b013e3181aaca87. Epub
[PubMed PMID: 19858696]
[19]
Bowne WB, Antonescu CR, Leung DH, Katz SC, Hawkins WG, Woodruff JM, Brennan MF, Lewis JJ. Dermatofibrosarcoma protuberans: A clinicopathologic analysis of patients treated and followed at a single institution. Cancer. 2000 Jun 15:88(12):2711-20
[PubMed PMID: 10870053]
[20]
Khatri VP, Galante JM, Bold RJ, Schneider PD, Ramsamooj R, Goodnight JE Jr. Dermatofibrosarcoma protuberans: reappraisal of wide local excision and impact of inadequate initial treatment. Annals of surgical oncology. 2003 Nov:10(9):1118-22
[PubMed PMID: 14597453]
[21]
Fields RC, Hameed M, Qin LX, Moraco N, Jia X, Maki RG, Singer S, Brennan MF. Dermatofibrosarcoma protuberans (DFSP): predictors of recurrence and the use of systemic therapy. Annals of surgical oncology. 2011 Feb:18(2):328-36. doi: 10.1245/s10434-010-1316-5. Epub 2010 Sep 16
[PubMed PMID: 20844969]
Level 2 (mid-level) evidence
[22]
Behbahani R, Patenotre P, Capon N, Martinot-Duquennoy V, Kulik JF, Piette F, Pellerin P. [To a margin reduction in the dermatofibrosarcoma protuberans? Retrospective study of 34 cases]. Annales de chirurgie plastique et esthetique. 2005 Jun:50(3):179-85; discussion 186-8
[PubMed PMID: 15935539]
Level 2 (mid-level) evidence
[23]
Veronese F, Boggio P, Tiberio R, Gattoni M, Fava P, Caliendo V, Colombo E, Savoia P. Wide local excision vs. Mohs Tübingen technique in the treatment of dermatofibrosarcoma protuberans: a two-centre retrospective study and literature review. Journal of the European Academy of Dermatology and Venereology : JEADV. 2017 Dec:31(12):2069-2076. doi: 10.1111/jdv.14378. Epub 2017 Jul 3
[PubMed PMID: 28573714]
Level 2 (mid-level) evidence
[24]
Lowe GC, Onajin O, Baum CL, Otley CC, Arpey CJ, Roenigk RK, Brewer JD. A Comparison of Mohs Micrographic Surgery and Wide Local Excision for Treatment of Dermatofibrosarcoma Protuberans With Long-Term Follow-up: The Mayo Clinic Experience. Dermatologic surgery : official publication for American Society for Dermatologic Surgery [et al.]. 2017 Jan:43(1):98-106. doi: 10.1097/DSS.0000000000000910. Epub
[PubMed PMID: 27749444]
[25]
Bichakjian CK, Olencki T, Alam M, Andersen JS, Berg D, Bowen GM, Cheney RT, Daniels GA, Glass LF, Grekin RC, Grossman K, Ho AL, Lewis KD, Lydiatt DD, Morrison WH, Nehal KS, Nelson KC, Nghiem P, Perlis CS, Shaha AR, Thorstad WL, Tuli M, Urist MM, Wang TS, Werchniak AE, Wong SL, Zic JA, McMillian N, Hoffman K, Ho M. Dermatofibrosarcoma protuberans, version 1.2014. Journal of the National Comprehensive Cancer Network : JNCCN. 2014 Jun:12(6):863-8
[PubMed PMID: 24925197]
[26]
Dagan R, Morris CG, Zlotecki RA, Scarborough MT, Mendenhall WM. Radiotherapy in the treatment of dermatofibrosarcoma protuberans. American journal of clinical oncology. 2005 Dec:28(6):537-9
[PubMed PMID: 16317260]
[27]
Navarrete-Dechent C, Mori S, Barker CA, Dickson MA, Nehal KS. Imatinib Treatment for Locally Advanced or Metastatic Dermatofibrosarcoma Protuberans: A Systematic Review. JAMA dermatology. 2019 Mar 1:155(3):361-369. doi: 10.1001/jamadermatol.2018.4940. Epub
[PubMed PMID: 30601909]
Level 1 (high-level) evidence
[28]
Amavi AK, Dossouvi T, Padaro E, Adabra K, Dosseh ED. [Management for locally advanced dermatofibrosarcoma protuberans in Togo]. Bulletin du cancer. 2018 Apr:105(4):333-334. doi: 10.1016/j.bulcan.2018.01.011. Epub 2018 Mar 2
[PubMed PMID: 29502795]
[29]
Yadav S, Verma N, Khurana N, Neogi S. Recurrent Dermatofibrosarcoma Protuberans with Pigmentation and Myoid Differentiation. Sultan Qaboos University medical journal. 2018 May:18(2):e228-e230. doi: 10.18295/squmj.2018.18.02.019. Epub 2018 Sep 9
[PubMed PMID: 30210857]
[30]
Asilian A, Honarjou N, Faghihi G, Saber M, Mozafarpoor S, Hafezi H. An experience of slow-Mohs micrographic surgery for the treatment of Dermatofibrosarcoma protuberans: A long-term cohort study. Journal of cosmetic dermatology. 2020 Oct:19(10):2701-2705. doi: 10.1111/jocd.13319. Epub 2020 Feb 10
[PubMed PMID: 32039548]