Continuing Education Activity
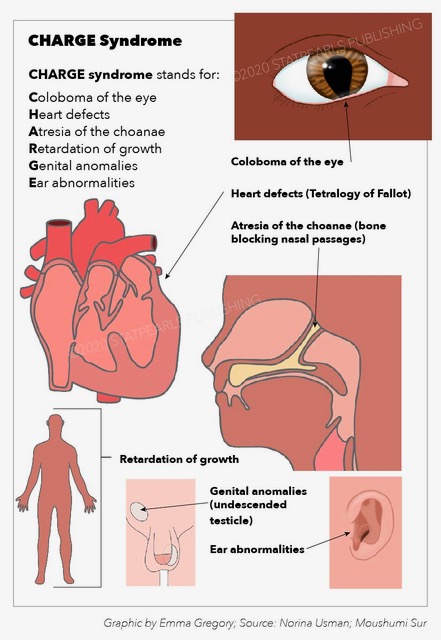
CHARGE syndrome is an inherited disorder caused by a mutation in the DNA-binding protein-7 CHD7 gene. CHARGE syndrome is an acronym for coloboma, heart disease, atresia of the choanae, retarded growth and mental development, genital anomalies, and ear malformations and hearing loss. CHARGE syndrome can have high morbidity, but the morbidity can be minimized by early diagnosis and treatment. This activity reviews the pathophysiology, evaluation, and treatment of CHARGE syndrome and highlights the role of the interprofessional team in managing patients and counseling families of patients with this syndrome.
Objectives:
- Explain the etiology and describe the pathophysiology of CHARGE syndrome.
- Outline the typical presentation and most common physical examination findings associated with CHARGE syndrome.
- Summarize the important aspects of medical and surgical management of CHARGE syndrome, including occupational, physical, and speech therapy.
- Review the importance of collaboration and communication amongst the interprofessional team to enhance the delivery of care for patients affected by CHARGE syndrome.
Introduction
CHARGE syndrome is a rare, autosomal dominant genetic disorder commonly diagnosed during the prenatal or neonatal period due to the identification of numerous dysmorphic and congenital anomalies. The features of CHARGE syndrome were first described independently by Hall and Hittner, and hence, it was initially called Hall-Hittner syndrome. Pagon first described the cardinal anomalies such as coloboma, choanal atresia, heart defects, genital abnormalities, retarded growth, and ear malformation and coined the term CHARGE syndrome. Diagnosis is now made using criteria proposed by Blake et al.[1] and further modified by Verloes,[2] who highlighted the importance of the 3C triad (coloboma, choanal atresia, abnormal semicircular canals). Other criteria include orofacial cleft, distinctive facial appearance, tracheoesophageal fistula, limb abnormalities, and rarely, immune deficiencies.
Mutations in the CHD7 (chromodomain helicase DNA binding protein) gene present on 8q12 results in several structural and physiological abnormalities, including hearing and vision loss, heart defects, and gastrointestinal (GI) problems. Among these, GI problems have a deep influence through an individual's life, with a subsequent increase in morbidity and mortality.
Etiology
Mutations in the CHD7 gene is the most common cause of CHARGE syndrome. In the rare case, it is autosomal dominant inheritance, which means it is transmitted from parent to child. Most cases arise from de novo mutations, which means that they are not inherited but occur sporadically. Therefore, abnormalities of the CHD7 gene, which usually occur de novo, can be found in over 90% of all children who meet the clinical diagnostic criteria.
The CHD7 gene controls the creation of a protein that regulates developmental pathways through chromatin organization. Mutations in the CHD7 gene lead to the making of an abnormally nonfunctional, short CHD7 protein, which interrupts the regulation of gene expression and disordered neural crest development. If these changes occur during the embryonic period, it leads to the development of the typically pleiotropic signs and symptoms of CHARGE syndrome.[3]
Epidemiology
CHARGE syndrome is a congenital disorder with an incidence of approximately 1 in 10000 births. It does not have sex-linked expression, so both males and females are equally affected. Most cases are sporadic and not inherited, but mean paternal age appears to be slightly increased. In 2010, a study on 379 patients diagnosed with CHARGE syndrome was conducted, followed by CHD7 mutation testing, and the result proved that 67% of cases were due to a CHD7 mutation.[4] There is a low recurrence rate of 1%.
Pathophysiology
Neural crest cells play an essential role in giving rise to its derivatives, including bone, pigment, smooth muscle, endocrine cells, neurons, glia as well as cartilage. Anomalies in neural crest-derived tissues can cause CHARGE syndrome. Deficiency in the passage of cervical neural crest cells into the pharyngeal pouches and arches, lack of mesoderm development, the defective interface between neural crest cells and mesoderm can subsequently lead to multiple phenotypic abnormalities. Mutations in the DNA-binding protein 7 (CHD7) gene cause CHARGE syndrome, as CHD7 plays a vital role in neural crest development.[5]
History and Physical
CHARGE syndrome is a pleiotropic disorder, including coloboma, heart defects, choanal atresia, retarded growth, genital abnormalities, ear anomalies, and deafness. A constant feature in CHARGE syndrome is semicircular canal hypoplasia ensuing in vestibular areflexia.[6] Other associated congenital disabilities involve bilateral or unilateral coloboma of the iris, bilateral or unilateral choanal stenosis or atresia. On physical examination, other abnormalities such as cranial nerve abnormalities, including anosmia, facial palsy (bilateral or unilateral), impaired hearing, and difficulty in swallowing, have been observed. Other features in CHARGE syndrome are abnormal ear shape, cryptorchidism, hypogonadism in both females and males, developmental delay, orofacial clefts, and tracheoesophageal fistula. Specific behavioral problems, such as autistic-like behavior, have been defined as well.[7][8]
A detailed history and a consideration of prenatal, neonatal, and childhood presentations are required. The prenatal performance needs to be assessed when poor fetal development, intrauterine growth restriction (IUGR), cleft lip or cleft palate, or a congenital heart defect is found in utero.
Neonates with CHARGE syndrome have multiple life-threatening medical conditions like small for gestational age, unable to pass nasogastric tube due to choanal atresia, dysmorphic features, and respiratory distress. Feeding difficulties are a significant cause of morbidity in all age groups.
Evaluation
The diagnosis of CHARGE syndrome is initially based on temporal bone imaging and clinical findings. The only gene associated with CHARGE syndrome is CHD7, encrypting the chromodomain helicase DNA binding protein. This sequencing detects pathogenic variants in maximum individuals with typical CHARGE syndrome with the following criteria of having the three primary characteristics or four major and three minor characteristics. The major criteria are the 4 C's: coloboma, cranial nerve abnormalities, choanal atresia, and typical CHARGE ear. The minor criteria are heart defects, cleft lip or palate, genital abnormalities, hypotonia, kidney abnormalities, esophageal atresia, poor growth, typical CHARGE face, and typical CHARGE hand.
In general, CHD7 analysis in individuals with either fewer major components or typical CHARGE syndrome detects pathogenic variants in around 65%-70% of cases.[9] The outline of diagnosis is summarized as clinical presentation, laboratory evaluation, genetic analysis, and imaging studies:[9]
- Clinical diagnosis: It is a combination of major and minor diagnostic characteristics, having the three primary features or four major and three minor characteristics.
- Laboratory analysis: It includes having the blood workup done, such as complete blood count (CBC), serum electrolytes, renal function test, luteinizing hormone-releasing hormone, Human chorionic gonadotropin (hCG), blood urea nitrogen (BUN), creatinine, growth hormone levels, and immunologic studies.
- Genetic analysis: Prenatal screening for CHD7 variants is restricted to familial cases, via amniocentesis chorionic or villus sampling at 10–12 and 18–20 weeks’ gestation.
- Imaging studies: Involves a skeletal survey, abdominal ultrasound, barium swallow, echocardiography, chest x-ray, cranial ultrasound in neonates, and head computed tomography (CT) scan and magnetic resonance imaging (MRI).
Treatment / Management
CHARGE syndrome patients need careful medical as well as surgical management strategies. Management is dependent on timing, features, and severity of the presentation. The most common neonatal emergencies in CHARGE syndrome include cyanosis due to congenital heart defects, or bilateral posterior choanal atresia, less likely due to trachea-esophageal fistula.[1]
- Therefore, all patients suspected of having CHARGE syndrome should have a cardiology consultation. If the infant has patent ductus arteriosus and limited pulmonary blood flow, prostaglandin should be given to maintain ductal patency.
- Some cases require tracheostomy to manage chronic airway difficulties, aspiration, or gastroesophageal reflux disease. Children with CHARGE syndrome need extensive medical management of their feeding difficulties, regularly needing jejunostomy or gastrostomy feeding tubes.[10]
- In children with CHARGE syndrome, intubation may be very difficult. Hence, a pediatric otolaryngologist or anesthesiologist should be present for planned surgical procedures.
- In patients with facial palsy, corneal scarring can be prevented by using artificial tears. Hearing aids should be used as quickly as hearing loss is recognized with regular followup.
- Regular remolding of the earpieces is required as the ear canals can be tiny initially, and ear cartilage may be inadequate to support a hearing aid. Cochlear implantations have been efficaciously done in CHARGE syndrome patients.[11] Children with CHARGE syndrome who experience cochlear implantation should be allowed to learn about sign language along with their communicative speech training.
- For endocrine issues, sex steroid therapy must be used for the descent of testes and penile growth in males with CHARGE syndrome. Testosterone is given for incomplete and delayed male puberty during adolescence. Females are given hormone replacement therapy if indicated at puberty. The use of sex hormone replacement can prevent osteoporosis.
- Assessment for cleft palate, together with submucous cleft palate and surgical correction at the appropriate age, must be done by an oral maxillofacial surgeon.
- Physical examination to evaluate the integrity of the cranial nerve in patients presenting with facial palsy.
- Ongoing and regular consultation with a genetic counselor or geneticist is recommended.[12]
Differential Diagnosis
- 2 deletion syndrome
- Isolated coloboma
- Isolated choanal atresia
- Isolated congenital heart defects
- Cat-eye syndrome
- Joubert spectrum
- Kabuki syndrome
- Holoprosencephaly spectrum disorders
- Renal coloboma syndrome
- VATER/VACTERL association
- Kalman syndrome
- Smith-Lemli-Opitz syndrome
- DiGeorge syndrome
Prognosis
Patients in the early years of life (infant age) are at high risk, specifically those infants who have severe congenital disabilities, resulting in poor outcomes and, thus, a high morbidity and mortality rate. Patients are most vulnerable in the first year of life and more prone to have an infection, require frequent hospitalization, and need complex surgeries, including congenital heart surgery. In late childhood through adulthood, more common causes of death include infection, aspiration, and obstructive sleep apnea. Widespread bilateral coloboma resulting in poor vision, brain malformation, microcephaly, lead to poor prognosis.[13]
Complications
- Swallowing difficulty
- Aspiration
- Corneal ulceration
- Retinal detachment
Deterrence and Patient Education
Families of patients with CHARGE syndrome need education about the disease manifestations and possible complications. Genetic counseling should be accessible for individuals at increased risk for potentially affected offspring to describe options in future pregnancies, including preimplantation and prenatal genetic diagnosis. Children with CHARGE syndrome can be assessed on a case by case basis. Some may require full-time support and individualized programs, and some may need little assistance. An essential step in dealing with abnormal behavior is understanding why it is occurring and helping the child learn more suitable methods of interaction. Before a child reaches the age of eighteen, care must be transitioned to adult clinicians and specialists that will manage the individual in later years.
Enhancing Healthcare Team Outcomes
An interprofessional team that offers a complete and unified approach to deliver services can accomplish the best results for a patient with CHARGE syndrome. Early identification of the syndrome and diagnostic workup to identify individual manifestations will help to prevent the associated complications. CHARGE syndrome is a heterogeneous condition, and consultation with a geneticist, pediatric cardiologist, pediatric otolaryngologist, or anesthesiologist is essential to make the shared decision that is essential for better outcomes. It is necessary to use a cohesive and coordinated care pathway based on evidence in the evaluation, planning, and management of all characteristics involved in the disease. The earlier a complication is recognized and managed, the better will be the prognosis.[14][15]