Continuing Education Activity
Hemophilia B is an inherited disease caused by a defect in the F9 gene, leading to insufficient production of the blood clotting factor IX. The genetic defect can occur either through X-linked inheritance or a spontaneous de novo mutation. Although this condition predominantly affects males, the carrier females may also experience occasional bleeding symptoms. Hemophilia B primarily follows an X-linked recessive pattern of inheritance. However, researchers have also reported some acquired forms resulting from the development of autoantibodies against factor IX. This topic comprehensively explores the genetic causes, clinical manifestations, potential complications, and the pivotal role of genetic testing in diagnosing hemophilia B, assessing disease severity, determining female carrier status, and informing decisions related to obstetrical care. This activity also highlights the role of the interprofessional team in evaluating and treating hemophilia B in individuals.
Objectives:
Identify genetic causes and risk factors associated with hemophilia B in patients.
Differentiate the clinical manifestations of severe, moderate, and mild hemophilia B through clinical assessment, laboratory testing, and genetic analysis.
Implement timely and appropriate diagnostic genetic testing for hemophilia B, including factor replacement therapy and prophylactic measures.
Collaborate with a multidisciplinary healthcare team, including hematologists, genetic counselors, dentists, and physical therapists, to provide comprehensive care for patients with hemophilia B.
Introduction
Hemophilia B, also known as Christmas disease, is the second most prevalent form of hemophilia. A defect in the F9 gene causes hemophilia B, leading to inadequate production of factor IX. The genetic defect can occur either through X-linked inheritance or a spontaneous de novo mutation. Although this condition predominantly affects males, the carrier females may also occasionally experience more significant bleeding symptoms. Heterozygous women with the F9 genetic mutation may have varying levels of factor IX, and those with levels at or above 50% of normal are usually asymptomatic.
Named after the first diagnosed case in 1952, Stephen Christmas, this disorder earned the moniker "the royal disease" due to its notable presence in the royal families of Spain, Germany, England, and Russia. Clinical presentations of the disease vary in severity, with males affected by the severe form displaying spontaneous and severe bleeding at birth. In contrast, individuals with milder cases usually experience bleeding primarily after trauma or surgery, and symptoms may not become apparent until later in life.[1][2][3] This topic comprehensively explores the genetic causes, clinical manifestations, potential complications, and the pivotal role of genetic testing in diagnosing hemophilia B, assessing disease severity, determining female carrier status, and informing decisions related to obstetrical care.
Etiology
Hemophilia A, B, and C result from deficiencies in clotting factors VIII, IX, and XI, respectively. The primary cause of these individual clotting factor issues is genetic, although sporadic cases are also possible. The genes for factors VIII and IX are present on the X chromosome. Hemophilia A and B are both inherited in an X-linked recessive pattern, and these conditions primarily affect males who can pass the gene to their daughters but not to their sons. Female carriers have a 50% chance of passing the gene to their offspring, and they are usually unaffected, as they possess one normal allele for the factor. Some females exhibit symptoms if they inherit mutated alleles from both parents or through the process of lyonization, where one X chromosome becomes inactivated.[1][2]
The development of antibodies against coagulation factors leads to acquired hemophilia B—an exceedingly rare form of hemophilia. Patients with acquired hemophilia typically possess a preexisting autoimmune disease, malignancy, or infection, such as HIV or hepatitis B or C.[3][4] The preferred terms for this form of hemophilia are "acquired factor inhibitor" or "acquired factor deficiency."
Epidemiology
Hemophilia occurs worldwide at a rate of 1 in 125,000. The prevalence of hemophilia B is 3.8 per 100,000 live males and 5 per 100,000 males at birth. The incidence is equal among all ethnic groups. Consanguinity can significantly contribute to frequency increases within specific communities.[5][6]
Pathophysiology
Injury to the vessel induces vasoconstriction by releasing endothelin and triggering a neural stimulation reflex. A disruption in the continuity of a vessel wall generates a series of cascades to create a hemostatic plug. The first step in the coagulation process involves the formation of a platelet plug, with exposed collagen enabling platelet secretion, adherence, and activation. The primary collagen receptors on platelets are the integrin glycoproteins GPIa/IIa and GPVI. Platelets secrete multiple substances from granules once they become activated. These substances recruit additional platelets, promote vasoconstriction, and serve as a source of von Willebrand factor (vWF) and fibrinogen during the clotting process.
After platelets are activated, the GPIIb/IIIa receptor on the platelet surface undergoes conformational changes, allowing them to bind with vWF and fibrinogen. Platelet aggregation occurs, leading to the development of large pseudopods and increased adhesiveness. The increased adhesiveness primarily occurs when the platelet surface receptor GPIb/IX/V complex binds to vWF. The binding of the platelet collagen receptor GPIa/IIa to collagen fibrils in the matrix enhances platelet adhesion.[7]
The clotting cascade activates, and the clot propagates. The formation of a fibrin mesh to stabilize the platelet plug requires the activation of 2 pathways—the intrinsic and extrinsic pathways. Collagen, basement membrane, activated platelets, and high molecular weight kininogen activate the intrinsic pathway, which comprises factors XII, XI, IX, and VIII. In contrast, tissue factor (TF) activates the extrinsic pathway, which includes factor VII. Activation of both extrinsic and intrinsic pathways leads to the activation of the combined pathway, consisting of factors X, V, II, and I, and forming a stable fibrin mesh. Hepatocytes synthesize factor IX, which is a part of the intrinsic pathway. A deficiency in factor IX leads to a defective coagulation cascade and inadequate formation of a fibrin mesh.[2]
The F9 gene associated with hemophilia B has multiple variants resulting from deletions, duplications, insertions, amino acid substitutions, or premature stop codons. Most known mutations consist of amino acid substitutions, also known as missense mutations. In some hemophilia B-affected families, the missense mutation interferes with factor activity but does not affect factor production. These patients will exhibit normal factor IX antigen levels but low factor activity during testing, which is referred to as cross-reacting material positive (CRM+).
Hemophilia B Leyden results from a rare mutation in the F9 promoter rather than the coding portion of the gene. This mutation affects the promoter region of the gene responsive to estrogen and testosterone, while leaving the promoter region unaffected by hormones. Puberty initiates the expression of some factor IX genes. As a result, individuals with hemophilia B Leyden may eventually develop factor IX levels at the lower end of the normal range.
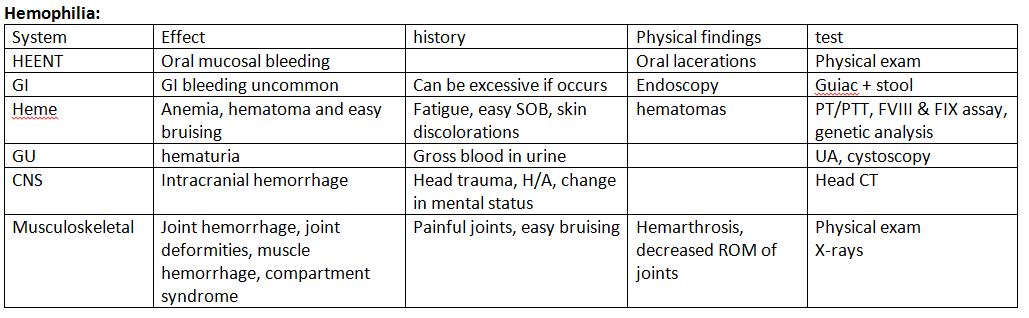
History and Physical
Clinically, hemophilia B is less severe than hemophilia A.[8] The degree of factor IX deficiency in the blood determines the presentation of disease manifestations.[9]
Severe Hemophilia
In severe hemophilia, bleeding occurs spontaneously and manifests early in life because coagulation factors are not transmitted transplacentally. Affected patients have less than 1% factor activity in their blood. During the newborn and infant period, symptoms may occur after circumcision, intramuscular vaccinations, mouth injuries, or when they begin to walk or crawl. As the patient ages, the central nervous system, oral or gastrointestinal tract, joints, and muscles become common bleeding sites.
Patients with severe hemophilia are at risk of developing organ bleeding in areas such as the liver, spleen, bladder, kidneys, and spinal cord. Intracranial hemorrhage (ICH) is the most life-threatening manifestation that occurs in 3% to 4% of patients with hemophilia at birth. In the newborn, ICH presents as seizures, hypotonia, focal weakness, apnea, or poor feeding. As individuals age, they may experience symptoms of ICH, such as headache, vomiting, and lethargy. However, the symptoms may remain asymptomatic and require detection through routine imaging. Extracranial bleeds, such as subgaleal bleeding and a cephalohematoma, can also be the initial presentation following delivery.[10]
Moderate Hemophilia
Patients with moderate hemophilia possess active factor levels ranging from ≥1% to ≤5%. In this condition, bleeding typically occurs after trauma, injury, dental work, or surgery in late childhood or adulthood. Furthermore, up to 25% of cases may experience recurrent joint bleeding.
Mild Hemophilia
Patients with mild hemophilia exhibit factor IX activity ranging from >5% to <40%. In this condition, bleeding occurs only after significant trauma or surgery. Spontaneous bleeding is uncommon, and the diagnosis is typically incidental.
In hemophilia B, hemarthrosis typically occurs in severe cases and represents the hallmark clinical presentation, accounting for nearly 80% of all hemorrhages related to hemophilia. The affected joints swell, become inflamed, cause pain, and feel warm, with limited range of movement. Once a joint sustains damage, it increases the risk of future bleeding events. Recurrent hemarthrosis eventually leads to the erosion of joint cartilage and the development of Charcot joints in hemophilic arthropathy. The knees, elbows, ankles, shoulders, wrists, and hips are the most commonly affected joints.[9] Hematomas in large muscle groups commonly present as another symptom. Repeated bleeding into muscles or bones can result in the formation of pseudotumors.
Patients may experience bleeding from the nose, oral mucosa, gingiva, and frenulum after dental procedures or trauma. Hematomas of the bowel wall can manifest as appendicitis or other acute abdominal pathology, leading to obstruction or intussusception. In addition, hematuria is a common occurrence.
Evaluation
A family history of hemophilia is present in approximately two-thirds of cases. Patients with a known family history of hemophilia or bleeding that exceeds expectations after a traumatic injury should undergo coagulation studies. Healthcare providers should evaluate the prothrombin time (PT), activated partial thromboplastin time (aPTT), and platelet levels of patients suspected of hemophilia.
Patients with hemophilia B will exhibit a prolonged aPTT, a normal PT, and normal platelet levels, indicating an intrinsic pathway disruption. As pregnancy and stress can falsely increase factor IX activity levels, it is essential to recheck the activity levels, if necessary, once these situations have been resolved. Patients with mild factor deficiencies may exhibit a normal aPTT. Healthcare providers perform mixing studies on blood from patients with an isolated prolonged aPTT to distinguish between a factor deficiency and an inhibitor.
Healthcare providers should conduct factor activity tests on males with a family history of hemophilia, patients without a known family history but with a clinical history consistent with hemophilia, and females who are known or may be genetic carriers. A factor IX activity level below 40% confirms the diagnosis of hemophilia B. Healthcare providers should consider genetic testing for all patients with hemophilia B as they assist in predicting which patients are likely to develop inhibitors and identifying carrier females in the family. An inhibitor is an antibody that develops against an infused factor and hinders its proper functioning. For patients with a confirmed diagnosis of hemophilia B, periodic laboratory evaluation involves screening for the presence of factor IX antibodies and testing for transfusion-related infections such as hepatitis and HIV.
Treatment / Management
When a female carrier is expecting a male infant, healthcare professionals must be prepared to handle the possibility of maternal and fetal bleeding. To avoid complications, they should avoid using fetal scalp electrodes and refrain from using forceps or vacuum assistance during delivery. Healthcare professionals should use the smallest possible needle for newborn screening, immunizations, and vitamin K administration. After the procedure, they should apply pressure to the site, use ice for 5 minutes, and delay circumcision until they confirm the diagnosis and assess factor activity levels. Healthcare professionals should administer routine immunizations on schedule to patients with hemophilia. Healthcare professionals should again use the smallest needle possible, restrict injections to one per limb, and apply pressure and ice for at least 5 minutes after the immunization. Patients who receive supplemental factors should get their vaccine when their factor levels are high.
Healthcare professionals consider regular exercise an integral part of health maintenance and prevention for patients with hemophilia. They recommend non-contact sports as the most appropriate options, which include swimming, walking, golf, tennis, bicycling, archery, and table tennis. Patients receiving supplemental factors and wearing suitable protective equipment can contemplate engaging in higher-impact sports. The decision to participate is individualized and made in conjunction with the healthcare team.
Patients should refrain from using aspirin, nonsteroidal anti-inflammatory drugs, and anticoagulants. Healthcare professionals should also engage in discussions with patients regarding over-the-counter herbal remedies and supplements, such as fish oil, which can heighten the risk of bleeding. Patients with heart disease may need aspirin or an anticoagulant. The healthcare team determines the appropriateness of these medications based on their risks and benefits. When traveling, patients should wear a medical alert bracelet, carry a supply of factor replacement, and be aware of the location of the nearest hemophilia treatment center if available.
Prophylaxis
The primary goal of factor IX prophylaxis is to enhance the quality of life by reducing hemarthrosis episodes, preventing the progression into hemophilic arthropathy, and minimizing episodes of intracerebral and muscular bleeds. Prophylactic treatment has limitations, including cost, inhibitor formation, product half-life, and the need for repeat venous access. Patients with severe hemophilia should receive prophylaxis regardless of bleeding episodes. Patients with 2 or more bleeding episodes also require prophylaxis. In patients with mild-to-moderate hemophilia and no prior bleeding episodes, prophylaxis is individualized based on their physical activity level, and these patients may consider intermittent prophylaxis.
Although prophylaxis offers several product options, the choice of agent should be individualized based on access and lifestyle.[11][12] The options include plasma-derived factor IX concentrate, recombinant factor IX, and longer-lasting recombinant factor IX. The process involves inserting a genetically engineered human factor IX gene into a Chinese hamster ovary cell line to create recombinant factor IX. Using this engineered product eliminates the issue of infectious complications. Fusing factor IX with a monomeric human immunoglobulin Fc domain (rFIXFc), polyethylene glycol, or the gene for albumin extends the half-life of factor IX. Trials have shown that the recombinant factor IX-Fc fusion protein reduces pain in hemophilia B, raises physical activity levels, and improves the quality of life.[13]
In 2022, the U.S. Food and Drug Administration (FDA) approved Etranacogene dezaparovovec—an adeno-associated virus 5 (AAV5) vector containing the F9 Padua variant.[14][15] This variant of the F9 gene contains a missense mutation that significantly increases F9 activity by 4- to 40-fold. Etranacogene dezaparovovec offers a one-time treatment option for adults with hemophilia B who use factor IX prophylaxis but still experience severe bleeding.
Management of Acute Bleeding
Replacing factor IX continues to be the primary treatment for patients with hemophilia B, with the dose determined by the severity of bleeding. The goal is to achieve 30% factor activity in patients with mild hemorrhages, 50% in patients with severe bleeding after trauma or those who need prophylaxis before major dental surgery or other surgery, and 80% to 100% in patients with life-threatening conditions.
The following formula calculates the appropriate dose:
The initial dose for factor IX = (patient's body weight (in kg)) x (desired factor IX increase (expressed as % in whole number)) x (factor accounting for the volume of redistribution (IU/kg; usually around 1 for factor IX)).[16]
For instance, a 45-year-old female with a body weight of 50 kg needs to increase her factor IX level by 100%, which is calculated as 50 x 100 x 1 = 5000 IU of factor IX.
Several strategies and treatment options can be utilized when managing bleeding episodes and dental extractions in patients with hemophilia, as mentioned below.
- Repeating the dose based on the half-life of the infused product.
- Considering prothrombin complex concentrate if factor IX is unavailable.
- Utilizing antifibrinolytic agents, including tranexamic acid and epsilon-aminocaproic acid, and monoclonal antibodies, such as rituximab, for consideration in cases of mucosal bleeds and dental extractions in patients with hemophilia.
Inhibitor Development
A significant complication in patients receiving factor IX replacement is the development of IgG antibodies that block the activity of the replaced factor. These inhibitory antibodies develop in response to exogenous factors and affect approximately 3% to 5% of patients with severe hemophilia B. Inhibitors occur much less frequently in patients with mild-to-moderate disease because the body does not recognize the infused factor as a foreign protein. Inhibitors complicate bleeding episodes by reducing responsiveness to factor infusions.[17] Healthcare providers should suspect inhibitors if bleeding does not cease after clotting factor replacement in a previously responsive patient. Anaphylactic reactions may occur with factor IX inhibitors.
Healthcare providers may consider alternatives such as plasmapheresis, bypassing products, or high-dose factor infusions for patients with hemophilia A and B who develop inhibitors. Recombinant activated FVIIa contains an activated form of a downstream clotting factor in the coagulation cascade. FVIIa can activate factor X independently of VIII or IX, but it can increase the risk of thrombosis, which varies from 1% to 10%.[18] Plasmapheresis is primarily used to treat individuals with life- or limb-threatening bleeding and may benefit patients with a high titer of an inhibitor. Plasmapheresis can acutely reduce the inhibitor titer, enabling transient use of replacement factor. Another option for individuals with bleeding who have developed an inhibitor is high-dose factor infusions.
Planning for Surgery
The surgical, anesthesiology, and hematology teams must collaborate with the lab and transfusion services for elective surgery. Pre-planning enables healthcare professionals to administer factors at predetermined intervals, monitor levels adequately, and determine the appropriate factor levels.
Future Treatment Considerations
Novel treatments such as gene therapy involve introducing exogenous DNA into a person's cells to produce a missing protein.[19] Researchers are currently developing cellular therapy, which involves subjecting cells to gene therapy outside of the body and then reintroducing them into the patient's body. They are also working on techniques to extend the half-life of factors and create factor alternatives. Monoclonal antibodies such as concizumab inhibit the coagulation cascade by targeting the TF pathway inhibitor (TFPI). This inhibition enables the generation of factor Xa and, subsequently, thrombin, even without factors VIII and IX.
Differential Diagnosis
Other diseases that may present similarly with bleeding episodes are listed below.
- Coagulation factor deficiencies, such as hemophilia A (factor VIII) and C (factor XI), exhibit similar presentations. Differentiation among the 3 is achieved through coagulation factor assay studies and genetic testing. Hemophilia A and B are X-linked recessive, whereas hemophilia C is autosomal recessive.
- vWF deficiency is the most common internal bleeding deficiency, with a defect in platelet plug formation. This disease is an autosomal disorder.[20] Affected patients have an increased bleeding time, normal or increased partial thromboplastin time (PTT), and normal platelets.
- Quantitative or qualitative platelet dysfunctions generally manifest as mucocutaneous bleeding, unlike hemophilia. The diagnosis is aided by platelet aggregation studies or electron microscopy. Typical findings include an increased bleeding time and a decrease in platelet count. Platelet dysfunction disorders include immune thrombocytopenia, thrombotic thrombocytopenia, and hemolytic uremic syndrome.
- Disseminated intravascular coagulation results in thrombosis and hemorrhage. Manifestations include a decreased platelet count, increased PT and PTT levels, elevated fibrin degradation (D-dimer), and reduced fibrinogen levels.[21] Usually, a precipitating event such as sepsis, trauma, obstetric complications, acute pancreatitis, acute promylogenous leukemia, or a transfusion triggers the condition.
- Neonates and patients with prolonged antibiotic use can experience vitamin K deficiency. This deficiency manifests as increased PT and PTT, decreased factors II, VII, IX, and X, and proteins C and S, along with normal platelet counts.[22]
- Scurvy, a vitamin C deficiency, presents with swollen gums, perifollicular and subperiosteal hemorrhage, hemarthrosis, and poor wound healing.[23]
- Ehlers-Danlos syndrome results from a defect in collagen synthesis and mainly presents with mucosal bleeding, hyperextensible skin, and hypermobile joints.[24]
- Healthcare professionals must maintain a high index of suspicion and listen for inconsistencies in the history of trauma when identifying child abuse, which can be misidentified and confused with hemophilia. Additional signs of abuse include different stages of wound healing, malnourishment, subdural hematoma, retinal hemorrhage, and signs of sexual abuse such as sexually transmitted infections and urinary tract infections.[25]
Prognosis
American physician Judith Graham Pool, a Nobel Prize winner in 1964, extracted cryoprecipitate from plasma containing higher amounts of coagulation factors and made significant improvements for patients affected by hemophilia. Prior to 1970, the life expectancy for patients with hemophilia was 11 to 13 years. In 1970, researchers extracted the first coagulation factor from plasma, significantly improving the quality of life and the prognosis of patients with hemophilia. Before the development of coagulation factors, affected patients received whole blood or fresh frozen plasma.
By the beginning of 1980, patients who received factor replacement presented with HIV and hepatitis C infections. HIV and hepatitis infections had affected 80% to 85% of patients who received clotting factors by 1992. The development of new screening techniques improved the safety of plasma-derived products and reduced the risk of transmission to patients.
Currently, patients in developed countries can achieve a near-normal life span if they have access to early factor replacement and prophylaxis starting from age 1 to 2 in cases of severe hemophilia. In developing countries, inadequate health systems and limited health resources contribute to a mortality rate for patients with hemophilia that remains twice that of the average healthy male.[26] Overall, there are still significant disparities in care. In upper-middle-income countries, those born with hemophilia have a 64% lower chance of living a life of average duration and quality, which increases to 77% in middle-income countries and up to 93% in low-income countries.
Complications
The most common complications of hemophilia are recurrent hemarthrosis, intracerebral hemorrhage, HIV and HCV infection, and the development of factor inhibitors. Recurrent hemarthrosis causes synovial membrane inflammation and hypertrophy, ultimately resulting in destructive arthropathy. Intracerebral hemorrhages can lead to chronic neurological disability. HIV and HCV infection can potentially result in death and hepatocellular carcinoma. Other concerns include hypovolemic shock caused by iliopsoas muscle bleeding and airway compromise resulting from retropharyngeal bleeding. In addition, patients with hemophilia experience higher rates of psychosocial and functional impairments and depression than normal healthy individuals. Maturational delays are also reported in children.[27]
Deterrence and Patient Education
Hemophilia is a condition that impairs normal blood clotting, potentially leading to life-threatening issues. Patients with hemophilia lack specific factors in their blood required for clot formation. Patients with hemophilia A lack factor VIII, whereas those with hemophilia B lack factor IX. Common symptoms of hemophilia include experiencing easy bruising, bleeding more than expected after a dental or surgical procedure, or bleeding into a joint. Although there is no cure for hemophilia, patients have access to treatments. Some may undergo prophylactic factor replacement on a scheduled basis, whereas others receive treatment only when necessary for an injury or surgery. Patients with hemophilia should apply cold packs, immobilize, use splints, take acetaminophen, and use codeine for acute injuries and pain. Anticoagulants, aspirin, and nonsteroidal anti-inflammatory medications may lead to complications.[28]
Children and adults should receive routine vaccinations. Healthcare providers should take special precautions when administering injections, including using a 23-gauge needle and applying ice packs and pressure for 5 minutes. Furthermore, injections should be limited to 1 per limb. Patients receiving factor replacement should schedule vaccines when factor levels are at their highest. Healthcare professionals should encourage appropriate dental care, advocating for at least twice-daily brushing and flossing. They should also emphasize the benefits of regular physical activity for patients with hemophilia, particularly non-contact sports. Patients are encouraged to discuss participation in more intense contact sports with their healthcare teams.
No data shows a preference for either cesarean or vaginal delivery in pregnant women carrying a child diagnosed with hemophilia in utero. Healthcare providers should avoid using forceps or vacuum extraction because they increase the risk of intracranial bleeding and cephalohematoma. Moreover, it is advisable to postpone circumcision until confirming or excluding the diagnosis.[29]
Enhancing Healthcare Team Outcomes
The main goal in treating patients with hemophilia B is to improve their quality of life and life expectancy. Many parts of the world have designated hemophilia treatment centers available to provide specialized care. These centers include teams of experts specializing in pain management, genetics, hematology, immunology, and the musculoskeletal system. The team also includes laboratory specialists, pharmacists, social workers, and psychologists.[2] Patients with hemophilia can receive comprehensive care from a team of specialists, allowing them to monitor symptoms and seek prompt treatment, which can reduce the risk of morbidity and mortality.
Home therapies are another integral part of treatment. The patient and family should become comfortable with hemophilia management to provide immediate access to early treatment and decrease overall pain, dysfunction, disability, and hospital admissions.[30]