Continuing Education Activity
Nephrocalcinosis, a term first coined in 1934 by Fuller Albright, refers to augmented calcium content within the kidney which can be in the form of calcium oxalate or calcium phosphate. However, there have been suggestions that the definition of nephrocalcinosis should be limited to calcium phosphate deposition only and deposition of calcium oxalate should carry the name oxalosis. This activity reviews the cause and presentation of nephrocalcinosis and highlights the role of the interprofessional team in its management.
Objectives:
- Identify the etiology of nephrocalcinosis.
- Review the investigation of a patient with nephrocalcinosis..
- Outline the treatment and management options available for nephrocalcinosis.
- Describe some interprofessional team strategies for improving care coordination and outcomes in patients with nephrocalcinosis.
Introduction
Nephrocalcinosis, a term first coined in 1934 by Fuller Albright, refers to augmented calcium content within the kidney which can be in the form of calcium oxalate or calcium phosphate.[1] However, there have been suggestions that the definition of nephrocalcinosis should be limited to calcium phosphate deposition only and deposition of calcium oxalate should carry the name oxalosis.[2] The condition demonstrates characteristic deposition of calcium in the kidney parenchyma and tubules. It can be incidentally detected in a radiograph from a patient with normal kidney function or present with an acute or chronic kidney injury. Many conditions lead to this entity, and the overall renal prognosis depends on the underlying causative condition causing.
Etiology
Nephrocalcinosis refers to generalized calcium deposition in the kidney and does not include the focal calcium deposition associated with focal renal injury. It is caused by diseases that cause hypercalcemia, hyperphosphatemia, hypercalciuria, hyperphosphaturia, and hyperoxaluria.
Calcium phosphate crystal formation occurs in alkaline urine pH. It has associations with many diseases which operate through the underlying mechanisms.
- Hypercalciuria with hypercalcemia
- Hypercalciuria without hypercalcemia
- Hyper-phosphaturia with hyperphosphatemia
- Hyper-phosphaturia without hyperphosphatemia
1. Conditions causing both hypercalcemia and hypercalciuria
- Primary hyperparathyroidism
- Vitamin D therapy
- Sarcoidosis
- Milk-alkali syndrome
- Congenital hypothyroidism
2. Conditions causing hypercalciuria without hypercalcemia
- Distal renal tubular acidosis
- Medullary sponge kidney
- Neonatal nephrocalcinosis and loop diuretics
- Inherited tubulopathies
- Chronic hypokalemia
- Beta thalassemia
3. Conditions causing Hyper-phosphaturia and hyperphosphatemia
- Tumor lysis syndrome
- Oral sodium phosphate bowel preparations
4. Conditions causing Hyper-phosphaturia without hyperphosphatemia
- Inherited tubulopathies such as Dent disease, Lowe syndrome
- X-linked, autosomal dominant, and autosomal recessive hypophosphatemic rickets.
Epidemiology
Primary Hyperparathyroidism
Nephrocalcinosis and nephrolithiasis are the most common renal manifestations of primary hyperparathyroidism.[3] Nephrocalcinosis has been reported in up to 22% of cases of primary hyperparathyroidism.[4] Although parathyroid hormone stimulates the distal tubular reabsorption of calcium, the effect is overshadowed by the increase in filtered calcium thereby, increasing the urinary calcium excretion.
Sarcoidosis
Sarcoidosis is associated with hypercalcemia and hypercalciuria. Nephrocalcinosis is seen in up to 50% of cases with renal involvement.
Hypervitaminosis D
Vitamin D acts by increasing the intestinal absorption of calcium and also bone resorption, which results in hypercalcemia and hypercalciuria. Nephrocalcinosis is more likely to occur when activated vitamin D such as calcitriol in conjunction with phosphate supplements. This scenario is common in X-linked hypophosphatemic rickets.[5] Children with nephropathic cystinosis excrete large amounts of calcium and phosphate due to renal tubular Fanconi syndrome and also receive substantial supplements of phosphate and alkalinizing agents. In a study by Theodoropoulos et al., it was found that out of 41 children with nephropathic cystinosis, 18 had mild nephrocalcinosis and 8 had severe nephrocalcinosis. The authors also concluded that nephrocalcinosis in such situations is best managed by reducing the oral replacement of calcium, phosphate, vitamin D, and citrate.[6]
Distal Renal Tubular Acidosis
It is the most common cause of nephrocalcinosis due to hypercalciuria without hypercalcemia. It has associations with hypocitraturia, which promotes calcium precipitation in renal tubules. It also results in metabolic acidosis which causes increased buffering of acid by bone buffers with subsequent release of calcium and phosphate. In a study by Brenner et al., evaluating the radiological prevalence of nephrocalcinosis in various renal tubular acidosis, of the 92 patients studied, nephrocalcinosis was seen in up to 29% of patients who were predominantly those with distal renal tubular acidosis.[7]
Medullary Sponge Kidney
This condition is a disorder with dilated papillary collecting ducts, and stones are common. It is associated with hypercalciuria and hypocitraturia thereby, resulting in nephrocalcinosis.[8] In a study by Yendt et al., nephrocalcinosis was observed in up to 50% of cases of the medullary sponge kidney.[9]
Nephrocalcinosis Due To Loop Diuretic Abuse
There is a risk of nephrocalcinosis in patients taking high doses of loop diuretics for a prolonged period. In a study by Kim et al., 18 consecutive adult patients who habitually took furosemide for weight gain and edema were evaluated. The study found nephrocalcinosis in 15 patients that underwent exposure to a mean dose of 538 mg per day. The authors concluded that the risk of nephrocalcinosis correlates with the dose of furosemide taken.[10]
Hereditary Disorders Associated with Nephrocalcinosis
Some of these include X-linked hypercalciuric nephrolithiasis, X-linked hypophosphatemic rickets, hypomagnesemia-hypercalciuria syndrome, and Bartter syndrome.
X-linked hypercalciuric nephrolithiasis is also termed Dent disease and has associations with some mutations affecting the CLCN5 gene on the X chromosome that leads to inactivation of CLC-5 voltage-gated chloride channels. This results in a clinical syndrome affecting young boys and usually includes hypercalciuria, nephrocalcinosis, nephrolithiasis, hematuria, low molecular weight proteinuria, glycosuria, aminoaciduria, hypophosphatemia, renal failure, and rickets.[11]
Pathophysiology
Nephrocalcinosis falls into the following classifications:
Molecular or chemical: an increase in intracellular calcium concentration which is quantifiable but not microscopically or radiologically visible
Microscopic: Deposits visible by light microscopic examination of kidney biopsy specimen but not radiologically visible
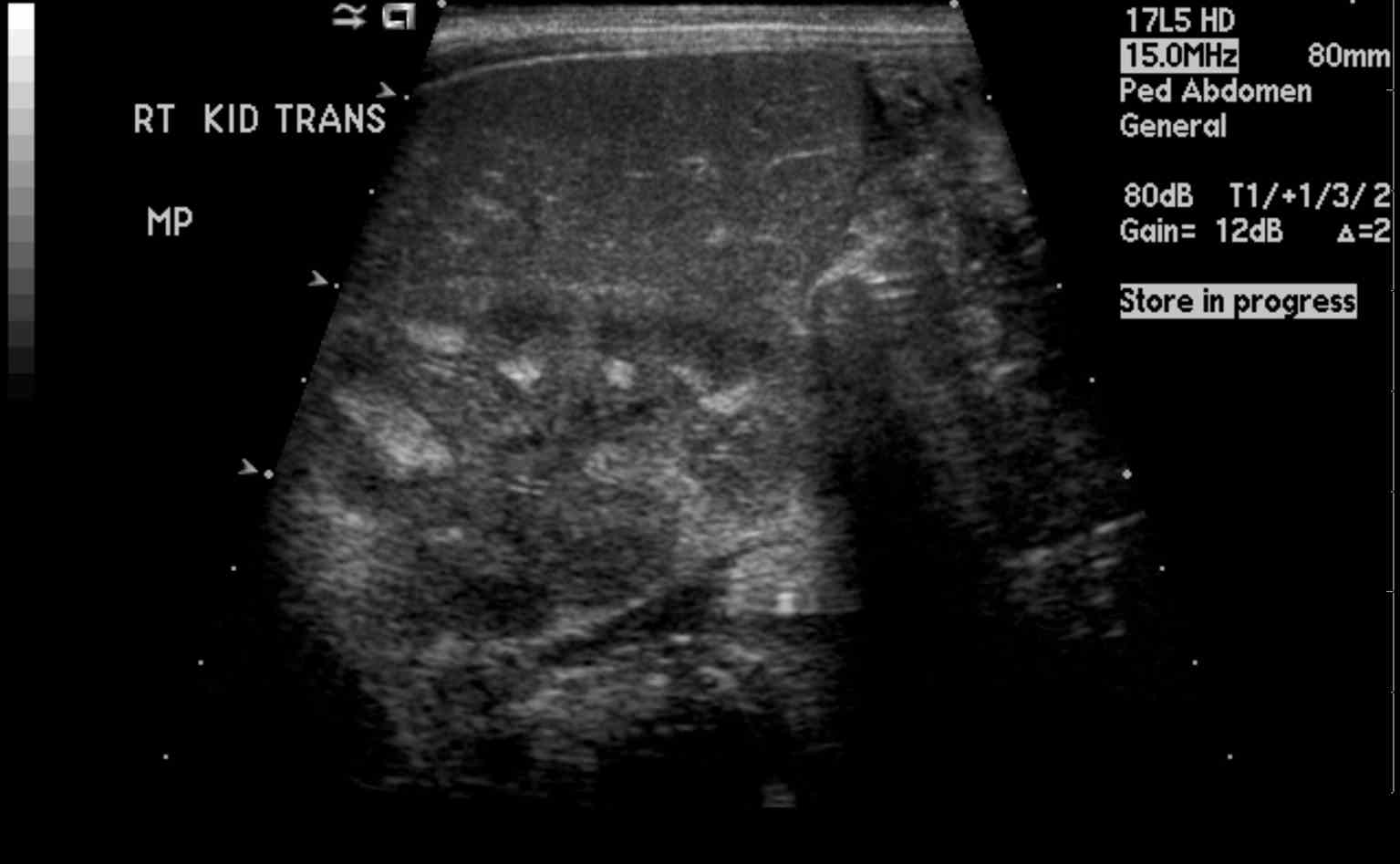
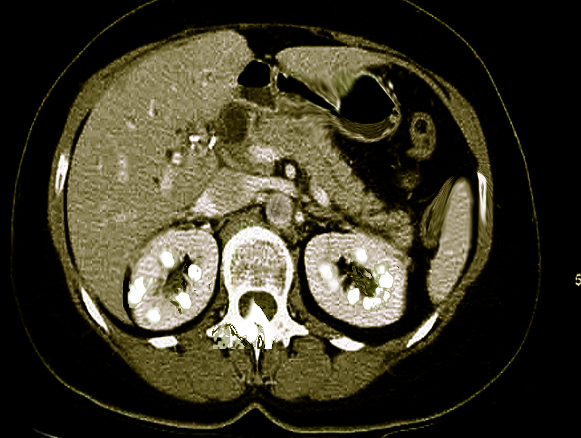
Macroscopic: calcification visible radiologically
It usually involves the renal medulla and much less often the cortex.
Conditions associated with cortical nephrocalcinosis are:
- Cortical necrosis such as occurs in pregnancy
- Transplant rejection
- Chronic glomerulonephritis
- Trauma
- Tuberculosis
- Oxalosis
Oxalosis is one condition which can cause both medullary and cortical nephrocalcinosis.
The most common cause of nephrocalcinosis is hypercalciuria with or without hypercalcemia. The metabolic abnormalities of hypercalciuria, hyperphosphaturia, and hyperoxaluria are often associated with nephrolithiasis and patients often present with both conditions. Calcium phosphate and calcium oxalate crystals form when the concentration of the solutes exceed the saturation limit in the tubular fluid. In a study by Evan AP et al., Randall's plaques were noted in the inner medullary interstitium as part of the basement membranes of the thin limb of the loop of Henle where the tubular fluid is saturated.[12] Once these calcium phosphate plaques form, they can enlarge into the surrounding interstitial tissue or rupture into the tubular lumen subsequently forming a nidus for luminal calcium oxalate stone.
Light microscopy in nephrocalcinosis shows intracellular, tubular, and interstitial basophilic calcifications. Calcium phosphate deposition is accompanied by lymphocyte predominant interstitial infiltrate and interstitial fibrosis.
History and Physical
Nephrocalcinosis most commonly presents as asymptomatic, chronic disease process discovered on radiographic imaging of abdomen. However, ordering radiographs might be feasible due to the evaluation of pain caused by coexisting nephrolithiasis. Nephrolithiasis can occur due to chronic hypercalcemia and hypercalciuria. It can cause renal colic. Polyuria and polydipsia can occur with chronic hypercalcemia, medullary nephrocalcinosis, and in Bartter syndrome.
It can also present acutely with renal failure in instances such as tumor lysis syndrome and acute phosphate nephropathy.
Evaluation
Once the radiologic determination of nephrocalcinosis has taken place, the causal investigation should follow. Serum electrolytes, calcium, phosphate should be measured. Measurement of urine pH should help determine the presence of distal renal tubular acidosis. Besides the above, the recommendation is to obtain two 24 hour urine collections for measuring the excretion of calcium, phosphate, oxalate, citrate, and creatinine.
Treatment / Management
Treatment is directed to treat the underlying cause of nephrocalcinosis. Strategies to reduce the urinary concentration of calcium, phosphate, or oxalate are used. Fluid intake is increased to produce at least 2 Liters of urine a day.
Among patients with hypercalciuria, urinary calcium excretion can be reduced by restricting the amount of animal protein, decreasing the sodium intake to less than 100 milliequivalents /day, increasing the potassium intake, and using a thiazide diuretic to reduce the urinary calcium excretion. However, patients with hypercalcemia should not use a thiazide diuretic.
In nephrocalcinosis due to distal renal tubular acidosis, potassium citrate is used in patients with hypocitraturia and pH less than 7. This not only replenishes the potassium loss but also achieves a normal urinary citrate level, thereby increasing the solubility of calcium.
Differential Diagnosis
Cortical nephrocalcinosis is typically the result of dystrophic calcification, which occurs subsequent to parenchymal tissue destruction rather than the precipitation of excessive urinary constituents. The causes include infarction, malignancy, and infection.
Prognosis
Specific causes of nephrocalcinosis such as Dent disease, primary hyperoxaluria, and hypomagnesemic hypercalciuric nephrocalcinosis, if not treated effectively, would progress to chronic renal failure,[13] End Stage Renal Disease. Nephrocalcinosis once detected radiologically is rarely reversible.
Complications
X-linked hypophosphatemic rickets if not treated appropriately will cause stunted bone growth. In hypomagnesemia-hypercalciuria syndrome children can present with tetanic seizures, muscle cramps, and weakness. In a subset of patients, sensorineural hearing loss and ocular impairment can occur.
Consultations
Nephrology
Urology
Pediatrics
Deterrence and Patient Education
Patients with nephrocalcinosis should be instructed to drink adequate water intake to maintain a urine volume of at least 2 liters/day. Patients need instruction to limit dietary protein intake in hypercalciuria, limit the daily sodium intake to less than 100 milliequivalents /day.
Enhancing Healthcare Team Outcomes
Knowledge of both the systemic and renal-specific causes of nephrocalcinosis is required as it will help to identify the primary lesion, whether it is genetic or acquired. It is vital to determine the underlying etiology for the nephrocalcinosis as only some causes will lead to chronic kidney disease and subsequently end-stage renal failure. Co-ordination between pediatrics, internal medicine and nephrology is needed to diagnose the primary etiology for early management.